
Ole Rasmus Theisen og Hilde Nordgarden
Medfødt tandmangel og ektodermale dysplasier
Tandlæge. TAKO-senteret, Lovisenberg Diakonale Sykehus, Oslo
Afdelingsleder, specialist i pædodonti, dr.odont. TAKOsenteret, Lovisenberg Diakonale Sykehus, Oslo
Klinisk relevans
Alvorlig medfødt tandmangel er en stor udfordring for patienterne, men også for tandplejeteamet. Især når tandmanglen er omfattende og indgår i en generel medicinsk tilstand som ektodermal dysplasi, er det yderst krævende at behandle og følge op på disse patienter. Der er behov for tværfaglig opfølgning og behandling, og patienterne skal følges fra tidlig alder.
Medfødt mangel af en enkelt eller nogle få tænder forekommer hyppigt i befolkningen, mens mangel af seks eller flere tænder (oligodonti) er relativt sjælden. Medfødt tandmangel kan være en isoleret tilstand eller indgå i et syndrom, hvoraf de forskellige ektodermale dysplasier er blandt de mest udbredte. Hypohidrotisk ektodermal dysplasi er den mest kendte og bedst beskrevne form for ektodermal dysplasi, og drenge med denne diagnose mangler i gennemsnit 22 permanente tænder. Desuden ses manifestationer fra andre væv som hår, hud og diverse kirtler, og dette kræver tværfaglig opfølgning og behandling. Tandplejeteamet har en vigtig og udfordrende rolle i både opfølgning og behandling af mennesker med ektodermale dysplasier i alle aldre.
Udtrykket hypodonti (fra græsk; hypo: mindre end normalt) bruges ofte ved medfødt mangel af en eller flere tænder, eksklusive tredjemolarer. Oligodonti betegner mere alvorlig tandmangel, mere specifikt medfødt mangel af seks eller flere tænder, eksklusive tredjemolarer. Anodonti betegner den mest alvorlige form for tandmangel, nemlig medfødt mangel af alle tænder i en kæbe [1]. I det primære tandsæt er hypodonti relativt sjælden [2]. Når den forekommer, er tilstanden ofte associeret med agenesi af den efterfølgende permanente tand [3]. I det permanente tandsæt er medfødt tandmangel derimod en af de hyppigst forekommende udviklingsforstyrrelser, som ses hos mennesker [4]. En metaanalyse, som inkluderede 33 studier, fandt, at prævalensen af medfødt tandmangel er højere i Europa (mænd 4,6 % og kvinder 6,3 %) og Australien (mænd 5,5 %, kvinder 7,6 %) end i Nordamerika (mænd 3,2 %, kvinder 4,6 %). Disse forskelle kan ifølge forfatterne skyldes forskelle i stikprøvestørrelse, upræcise observationer og diverse lokale ætiologiske faktorer [5].
Oligodonti er mere sjælden og har ifølge skandinaviske studier en prævalens på 0,08-0,16 % [6][7][8]. Oligodonti kan optræde som en isoleret tilstand eller som del af et syndrom [9]. Tilstanden kan være associeret til ændringer i tændernes størrelse (små tænder) og form (koniske tænderog taptænder), forsinket tandfrembrud, emaljeudviklingsforstyrrelser og forstyrrelser i kæbevæksten [10].
Hvis man søger i databasen OMIM® (Online Mendelian Inheritance in Man®) med søgeord, som dækker udtryk for medfødt tandmangel (hypodontia, oligodontia, tooth agenesis, missing teeth), får man over 1.500 hits på diverse genetiske diagnoser. Det er ikke muligt at beskrives alle disse i en oversigtsartikel.
Oligodonti er imidlertid særlig udtalt og udbredt ved forskellige former for ektodermal dysplasi, hvilket er vist i flere studier. I et hollandsk studie undersøgte man 167 patienter med oligodonti og fandt, at 53 % havde isoleret oligodonti, og 47 % havde oligodonti som del af et diagnosticeret eller mistænkt syndrom. Blandt de 48 patienter, som havde et påvist syndrom, havde 85,4 % en form for ektodermal dysplasi [11]. I et norsk studie, hvor man så på 68 patienter med oligodonti, havde 57 % også kliniske tegn på udviklingsforstyrrelser i andre ektodermale væv og blev klinisk diagnosticeret med ektodermal dysplasi [12].
Ektodermale dysplasier
Forstyrrelser i udviklingen af embryonal ektoderm kan føre til en række sygdomme/tilstande, herunder en heterogen gruppe diagnoser, som kaldes ektodermale dysplasier (ED). ED blev oprindeligt defineret som tilstande, hvor der er udviklingsforstyrrelser i to eller flere strukturer, som stammer fra embryonalt ektoderm, hovedsagelig strukturer, som udvikles fra huden og hudens vedhæng (svedkirtler, hår og negle) samt tænderne
[13][14]. I 2017 blev denne klassificering revideret af en international ekspertgruppe, som slog fast, at ektodermale dysplasier er genetiske tilstande, der påvirker udviklingen og/eller homeostasen af to eller flere ektodermale derivater, inklusive hår, tænder, negle og visse kirtler [15]. Mere end 180 forskellige typer ED er blevet beskrevet på baggrund af fænotype og eventuelt arvegang [14][16].
Nyere genetiske metoder gør, at man i dag oftere får stillet en mere nøjagtig diagnose, som definerer, hvilke specifikke gener og molekylære signalveje der er involveret i sygdomsudviklingen. I den seneste publikation om klassificering af ektodermale dysplasier fra en international ekspertgruppe er definitionen af ED igen blevet modificeret:
»Ektodermale dysplasier er medfødte, genetiske tilstande, som påvirker udviklingen af to eller flere ektodermale væv, inklusive hår, tænder, negle og visse kirtler.«
I denne nyeste publikation opregnes 49 forskellige former for ektodermale dysplasier, hvor den molekylære genetiske baggrund er kendt. En del diagnoser, som tidligere var inkluderet, er nu ikke længere med på listen, fordi der kun var beskrevet enkelttilfælde, eller fordi den molekylære baggrund ikke er kendt [17].
Hypohidrotisk ektodermal dysplasi
Den mest beskrevne og undersøgte form blandt de mange forskellige ektodermale dysplasier er hypohidrotisk ektodermal dysplasi (HED). Man antager, at omkring én ud af 15.000 personer har HED, men at nogle kan have tilstanden i så mild form, at den sandsynligvis er underdiagnosticeret. HED kan nedarves på flere måder; men de gener, som er involveret, har funktioner i en signalvej (ektodysplasin/NF- B), som er helt nødvendig for korrekt udvikling af flere ektodermale strukturer. Mutationer i genet EDA på X-kromosomet forårsager ca. 80 % af tilfældene af HED [18], og i disse tilfælde bruges betegnelsen X-bundet HED (XLHED). Piger med mutation i et af EDA-generne kaldes ofte heterozygote bærere, og de får sædvanligvis mildere symptomer. Drenge har kun ét X-kromosom, og mutation i EDA-genet fører derfor til mere udtalte symptomer.
Betegnelsen hypohidrotisk betyder, at diagnosen fører til reduceret svedsekretion. Personer, især drenge, med HED har derfor problemer med at regulere kropstemperaturen og har dermed risiko for overophedning. Som nyfødte kan de have uforklarlig feber (hypertermi), som i enkelte tilfælde kan føre til feberkramper og i sjældne tilfælde permanent hjerneskade. Dette er også en risiko ved feber som følge af andre sygdomme i løbet af barneårene [19]. Også andre kirtler end svedkirtler kan afficeres, fx tårekirtler, spytkirtler (omhandles mere detaljeret nedenfor), mælkekirtler og meibomske kirtler (kirtler, som tilfører lipider til øjet) [20]. Der kan også være reduceret antal kirtler i de øvre luftveje. Reduceret sekretion fra kirtler kan føre til problemer med tørre øjne, hud og mund, problemer med amning samt hyppige luftvejsinfektioner. Mennesker med HED har ofte tyndt, lyst og sparsomt hovedhår, som let knækker, og tynde eller manglende øjenbryn og -vipper [21]. Drenge med HED har typisk også karakteristiske ansigtstræk med en fremtrædende pande, tyndt og lyst hår på hovedet og tynde øjenbryn. De har ofte tør hud, især omkring øjnene, og fyldige læber (Fig. 1).

Fig. 1. Illustration af en ung mand med HED.
Illustration: Malin Bernas-Theisen, TAKO-senteret.
Manglende tænder ved hed
Medfødt tandmangel ses hyppigt ved HED. Et dansk studie, som inkluderede 23 mænd med XLHED og 36 heterozygote kvinder, fandt, at mændene i gennemsnit manglede 22 tænder (14-28) og kvinderne i gennemsnit fire tænder (0-22) [22]. De tænder, der hyppigst mangler, er laterale incisiver i overkæben, første præmolarer i begge kæber og underkæbeincisiver, mens de tænder, som oftest er til stede, er første centrale incisiver i overkæben, førstemolarer i overkæben, førstemolarer i underkæben og hjørnetænder i begge kæber [22]. Lignende agenesimønster ses ved ektodermale dysplasier generelt og hos patienter med isoleret oligodonti [12] (Fig. 2).
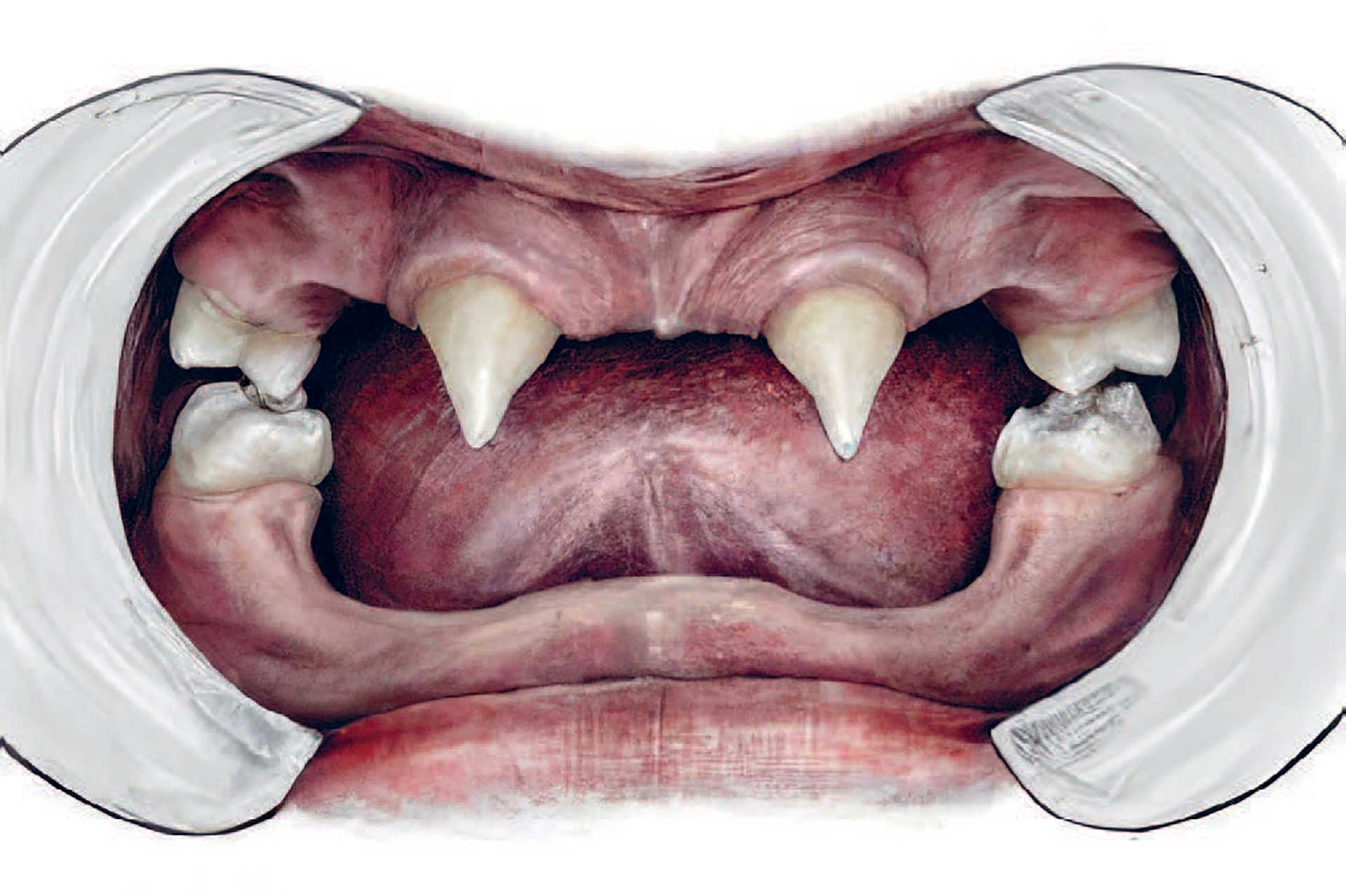
Fig. 2. Drenge med HED udvikler oftest få tænder, og tænderne har atypisk form. Illustration: Malin Bernas-Theisen, TAKO-senteret.
Tandform
Ved HED ses ofte, at tænderne har en karakteristisk form. Fortænderne er koniske og smallere end gennemsnittet (Fig. 2). Den koniske form er mest udtalt hos afficerede drenge; men heterozygote piger har også ofte ændret tandform, idet tænderne er smallere mod incisalkanterne (taptænder) [22]. Det danske studie viste også, at taurodonti hyppigt forekommer. Taurodontisme er en anomali, som opstår i tænder med flere rødder, især molarer. Pulpakammeret er forlænget mod rodspidsen, og den sædvanlige indsnævring, som ses ved emaljecementgrænsen, er fraværende (Fig. 3). Taurodonti kan være en udfordring, hvis der bliver behov for endodontisk behandling af afficerede tænder.
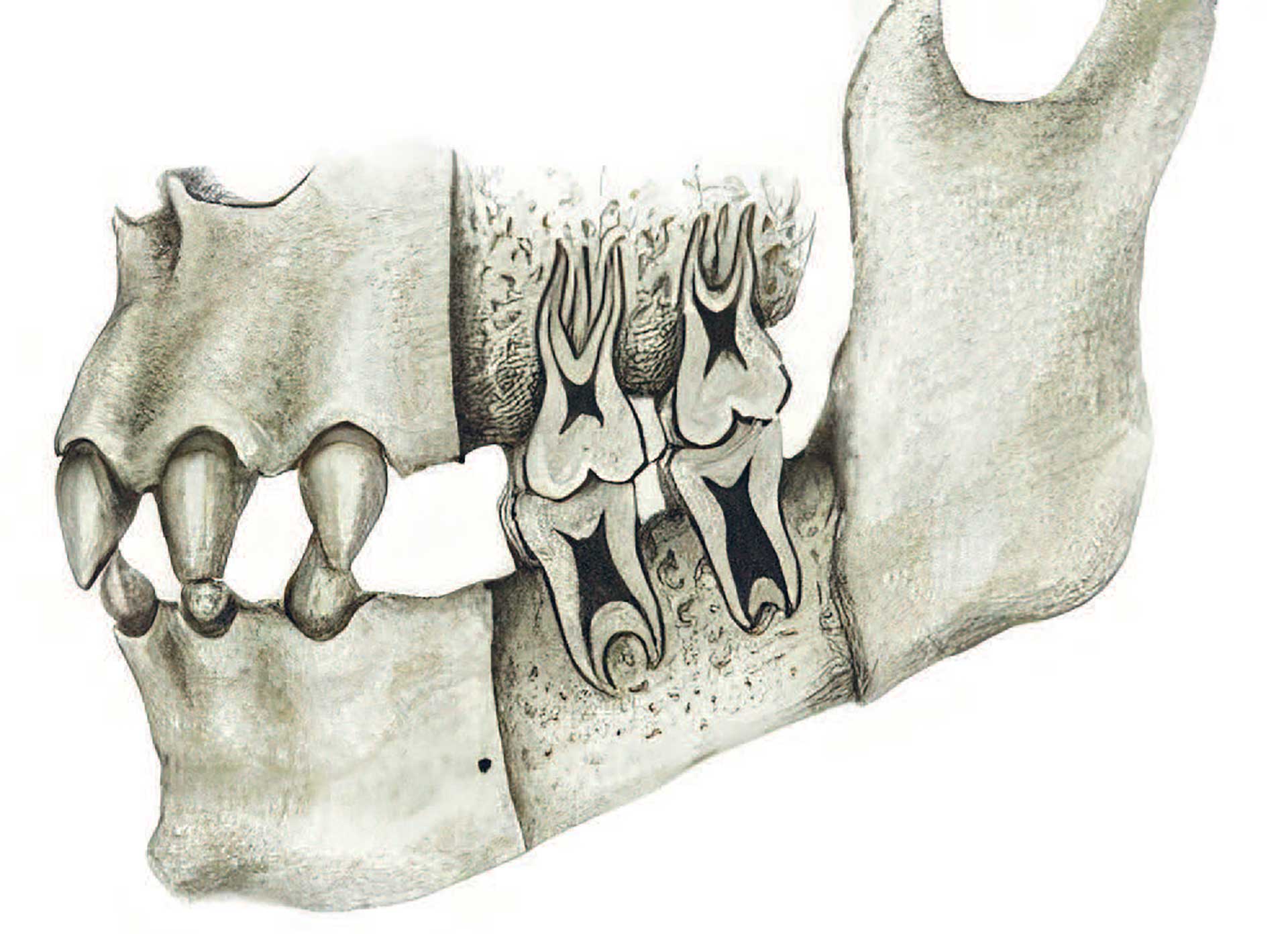
Fig. 3.Illustration af taurodonte molarer i en kæbe, der er afficeret af HED. Illustration: Malin Bernas-Theisen, TAKO-senteret.
Saliva
Spytkirtlerne er også ektodermale derivater og er ofte underudviklet ved forskellige former for ektodermal dysplasi. Det er rapporteret i flere studier, at mennesker med ektodermal dysplasi har lavere salivasekretion end raske kontrolpersoner [12][22], og dette kan føre til tørre og følsomme slimhinder. Trods dette er de subjektive gener i relation til reduceret salivasekretion relativt beskedne, måske fordi personer med HED aldrig har oplevet normal salivasekretion og dermed ikke giver udtryk for, at de føler sig tørre i munden [12]. Cariesrisikoen synes ikke at være højere hos personer med ektodermal dysplasi, faktisk har man i et studie fundet, at børn med ektodermal dysplasi havde meget lavere prævalens af caries end børn med amelogenesis imperfecta og raske kontroller [23].
Mund- og ansigtsfunktioner
Mund- og ansigtsfunktioner, også kaldet orofaciale funktioner, inkluderer tale, mimik, spisning, væskeindtagelse, rensning af mundhulen og håndtering af spyt. Reduceret salivasekretion og mange manglende tænder kan gøre det vanskeligt at spise og synke. Tyggeproblemer som følge af manglende tandkontakter forekommer ofte. Endvidere kan reduceret spytmængde gøre det vanskeligt at flytte mad rundt i munden og danne en bolus. Der er også rapporteret læspen og hæshed [24].
Præcisionsmedicin ved x-bundet hypohidrotisk ektodermal dysplasi
Persontilpasset medicin, eller præcisionsmedicin, er et begreb, som de senere år har vundet indpas. Det betyder skræddersyet behandling, som er tilpasset personens genetiske profil [25]. Der foregår spændende forskning inden for dette felt, når det drejer sig om X-bundet hypohidrotisk ektodermal dysplasi (XLHED). XLHED skyldes mutationer i genet EDA, som medfører mangel på proteinet ektodysplasin. Dette gen blev først beskrevet som årsag til XLHED i 1996 [26]. Allerede i 2003 kunne en forskergruppe korrigere fænotypen hos musefostre med denne diagnose ved at tilføre rekombinant fremstillet protein, som illuderer ektodysplasin, til cirkulationen [27] og senere til fostervandet [28] hos drægtige mus. I 2018 publicerede Schneider og medarbejdere [29], at de havde injiceret rekombinant protein i fostervandet på en mor, som bar på to drengefostre (tvillinger) med XLHED. Moderen havde i forvejen en søn med XLHED. Injektionen blev givet i uge 26 og gentaget i uge 31. Tvillingerne blev født raske i uge 33. Ved seksmånedersalderen havde begge tvillinger normal tæthed af svedkirtler og normal pilokarpininduceret svedsekretion, mens deres storebror med samme mutation slet ikke kunne svede. De to første somre, tvillingerne levede, havde de ingen episoder med hypertermi eller sygehusindlæggelser i forbindelse med reduceret lungefunktion. De producerede også normale mængder saliva. En anden gravid kvinde, som ligeledes havde en søn med XLHED, blev behandlet på samme måde i uge 26, efter at man ved ultralyd havde påvist mange manglende tandanlæg hos fosteret og bekræftet diagnosen med genetisk test. Drengen blev født rask i uge 39. Denne dreng fik også forøget tæthed af svedkirtler, men ikke i samme grad som tvillingerne. Han havde også noget mindre svedproduktion ved seksmånedersalderen, men normalt antal meibomske kirtler i øjenlågene. Radiologiske undersøgelser viste, at denne dreng havde ni tandanlæg i seksmånedersalderen, mens hans afficerede storebror kun havde to [29]. Et større fase 2-studie af denne behandling er nu iværksat [30]. I fremtiden kan man håbe og tro, at tilsvarende behandling vil blive udviklet for flere typer ektodermal dysplasi, og dette understreger betydningen af god diagnostik så tidligt som muligt.
Andre former for ektodermal dysplasi
Som nævnt er der aktuelt defineret 49 forskellige former for ektodermal dysplasi. Der er planer om regelmæssige opdateringer af denne liste, da forskningen stadig gør fremskridt med henblik på at identificere den genetiske baggrund og de molekylære signalveje, som ligger til grund. Af pladshensyn kan alle disse diagnoser ikke beskrives i detaljer her. Mange af diagnoserne har dog lignende symptomer og orale fund som beskrevet for HED. Det er dog ikke alle med ED, der mangler tænder, og nogle kan have andre udviklingsforstyrrelser som emaljehypoplasi og -hypomineralisering [31][32].
Diagnostik, opfølgning og behandling af personer med ED
Eftersom orale forandringer så hyppigt ses ved mange former for ED, kan det i mange tilfælde være tandplejepersonale, der først får mistanke om diagnosen. Der er i sådanne tilfælde international enighed om, at patienten skal henvises videre til genetisk undersøgelse for at bekræfte diagnosen og få genetisk vejledning [33].
Personer med ED kan have komplekse behov, og de har ofte behov for kontakt med mange forskellige fagpersoner som børnelæger, hudlæger, øre-næse-hals-læger, øjenlæger, genetikere og et tandplejeteam, som typisk inkluderer specialister i pædodonti, ortodonti, oral rehabilitering og oral kirurgi. Der kan også være behov for at trække på logopæder, psykologer, fysioterapeuter og andre medicinske faggrupper. Ved en international konference i 2004 [34] blev man enige om en række basale krav til opfølgning og behandling af personer med ektodermale dysplasier generelt
Alle patienter med ED bør have adgang til fagpersoner i den primære sundhedstjeneste, som har kendskab til diagnosen, så tilstanden kan blive korrekt diagnosticeret, og de kan henvises videre ved behov.
Patienternes sundhedskontakter bør arbejde tæt sammen med patienten og patientens omsorgspersoner.
ED og tandplejeteamet
En god og tillidsfuld relation til tandplejepersonalet er vigtig. Mange med ED får stort behov for tandbehandling over lang tid, og det kan opleves som psykisk belastende [35], hvilket man må tage hensyn til i behandlingsplanlægningen [33]. Børn med ED bør have en fast tandlæge med indsigt i diagnoserne, som kan informere om og koordinere behandlingsforløbet og inddrage nødvendig ekspertise efter behov.
Det er afgørende at forebygge tandsygdomme og bevare primære tænder uden permanente efterfølgere så længe som muligt, hvis rodmorfologi og stabilitet tillader det [33]. Hvis de primære tænder kan bevares, vil man kunne beholde en bedre kæbekam omkring tænderne, og dette vil kunne bidrage til at forenkle den endelige rehabilitering. Der er international konsensus om vigtigheden af, at patienter og pårørende informeres om mundhygiejne, at tandbørste og fluoridtandpasta bruges jævnligt, at cariesrisikoen vurderes individuelt i lyset af de medicinske forhold, at patienten har adgang til et tandplejeteam, og at man bør måle salivasekretionen hos patienter med ED [33].
Der er behov for en grundig undersøgelse af tænder og kæber hos personer med ED, selvsagt med fokus på de ovennævnte fund og symptomer. Det kan være nyttigt at undersøge de orofaciale funktioner ved hjælp af en valideret test, NOT-S [24]. Der er ingen retningslinjer for, hvornår der første gang bør tages panoramarøntgen af børn med ED, så dette må afhænge af behandlerens skøn i hvert enkelt tilfælde [36].
Restaurering af tænder med unormal form foretages med kompositmaterialer som førstevalg hos de yngste børn, mens man kan overveje porcelænsmaterialer hos ældre børn, unge og voksne [33].
Erstatning af manglende tænder er ofte krævende. Valget af behandling afhænger af flere faktorer såsom alder, udviklingstrin, anatomi af både blødt- og hårdtvæv og antallet af manglende tænder. Både aftagelige hel- eller delproteser, faste tandunderstøttede proteser, dækproteser og implantatunderstøttede proteser kan blive aktuelle [37].
Man har diskuteret, på hvilket alderstrin man bør igangsætte rehabilitering ved manglende tænder, og det er der flere holdninger til. I det allerede nævnte internationale studie [33] var der enighed om, at man ikke starter rehabilitering før toårsalderen, mens der var uenighed om anvendelse af aftagelig protetik hos børn fra 2-3-årsalderen. Aftagelig protetik er sjældent indiceret så tidligt; men det kan være psykologisk gavnligt, at børnene får tanderstatninger, inden de begynder i skolen [36]. Man bør være opmærksom på, at tørre slimhinder kan føre til dårlig retention af aftagelige proteser og gøre det svært for børnene at bruge disse [37].
De fleste med ektodermal dysplasi vil have et ønske om faste tanderstatninger, ofte implantater. Implantatunderstøttede proteser har været anvendt ved ED i mange år, også hos børn ned til fireårsalderen. Implantatbehandling bliver ifølge ét studie ofte vellykket hos børn, når implantaterne indsættesi underkæbefronten [37], mens et andet studie fandt, at ni ud af 14 implantater gik tabt før belastning hos børn mellem fem og 12 år [38]. Vellykket implantatbehandling afhænger af, om der er nok alveolarknogle af god kvalitet. Den vigtigste faktor til fastlæggelse af det ideelle tidspunkt for implantatindsættelse er patientens skeletale alder/modenhed. Man kan desuden overveje tidlig behandling med implantater i alvorlige tilfælde af tandmangel ved ED for at undgå dårlig oral funktion ved anvendelse af aftagelig protetik.
Det er ofte nødvendigt at foretage knogleopbygning for at forøge alveolarkammens vertikale højde og bredde i forbindelse med implantatindsættelse hos personer over 16 år med ektodermal dysplasi. I de fleste tilfælde må knogletransplantatet hentes fra andre dele af kroppen, typisk fra bækkenet [37]. Ved brug af zygomaticusforankrede implantater er det muligt at undgå knogletransplantation til overkæben. Disse har i små studier med opfølgningsperioder op til 12 år vist god prognose [39][40].
Studier af implantatbehandling hos både børn og voksne varierer betydeligt med henblik på implantattype, suprakonstruktion, design og opfølgningsperiode. Wang et al. (2016) [37] fandt i en oversigtsartikel, at 14 ud af 701 implantater, der blev indsat hos 96 børn, unge og voksne, måtte fjernes i løbet af en observationsperiode på to år.
Konklusioner
Oligodonti kan optræde som en isoleret tilstand eller som en del af en mere omfattende genetisk diagnose, hvoraf diverse former for ektodermal dysplasi er de hyppigste. Når tandplejeteamet møder patienter med oligodonti, er det yderst vigtigt, at de engagerer sig i et multidisciplinært samarbejde mkring diagnostik, behandling og behandlingsplanlægning. Adgang til molekylær genetisk diagnostik er vigtigt. En genetisk diagnose giver bedre forudsigelighed, adgang til genetisk rådgivning og fører i nogle tilfælde til, at man opdager behov for anden medicinsk opfølgning. Fx kan visse genmutationer, som fører til agenesi af tænder, også disponere for mere alvorlige tilstande som forskellige former for cancer ved AXIN2-mutationer [41]. Ved planlægning og gennemførelse af tandbehandling er det også vigtigt, ar der sker et samarbejde mellem flere specialister, og både timing og valg af behandling vil variere individuelt
Litteratur
Schalk-van der Weide Y, Steen WH, Bosman F. Distribution of missing teeth and tooth morphology in patients with oligodontia. ASDC J Dent Child. 1992;59:133-40.
Nieminen P. Genetic basis of tooth agenesis. J Exp Zool Mol Dev Evol. 2009;312B:320-42.
Bailleul-Forestier I, Molla M, Verloes A et al. The genetic basis of inherited anomalies of the teeth. Part 1: clinical and molecular aspects of non-syndromic dental disorders. Eur J Med Genet. 2008:51;273-91.
Matalova E, Fleischmannova J, Sharpe PT et al. Tooth agenesis: from molecular genetics to molecular dentistry. J Dent Res. 2008;87:617-23.
Polder BJ, Van’t Hof MA, van der Linden FP et al. A meta-analysis of the prevalence of dental agenesis of permanent teeth. Community Dent Oral Epidemiol. 2004;32:217-26.
Nordgarden H, Jensen JL, Storhaug K. Reported prevalence of congenitally missing teeth in two Norwegian counties. Community Dent Health. 2002;19:258-61.
Bergendal B, Norderyd J, Bågesund M et al. Signs and symptoms from ectodermal organs in young Swedish individuals with oligodontia. Int J Paediatr Dent. 2006;16:320-6.
Rølling S, Poulsen S. Oligodontia in Danish schoolchildren. Acta Odontol Scand. 2001;59:111-2.
Salinas CF, Irvine AD, Itin PH et al. Second International Conference on a classification of ectodermal dysplasias: development of a multiaxis model. Am J Med Genet A. 2014;164A:2482-9.
Schalk-van der Weide Y. Oligodontia. A clinical, radiographic and genetic evaluation. [PhD thesis] Utrecht: University of Utrrect, 1992.
Schalk-van der Weide Y, Beemer FA, Faber JA et al. Symptomatology of patients with oligodontia. J Oral Rehabil. 1994;21:247-61.
Nordgarden H, Jensen JL, Storhaug K. Oligodontia is associated with extra-oral ectodermal symptoms and low whole salivary flow rates. Oral Dis. 2001;7:226-32.
Freire-Maya N. Ectodermal dysplasias. Hum Hered. 1971;21:309-12.
Pagnan NAB, Visinoni AF. Update on ectodermal dysplasias clinical classification. Am J Med Genet A. 2014;164A:2415-23.
Wright JT, Fete M, Schneider H et al. Ectodermal dysplasias: Classification and organization by phenotype, genotype and molecular pathway. Am J Med Genet A. 2019;179:442-7.
Itin PH. Etiology and pathogenesis of ectodermal dysplasias. Am J Med Genet A. 2014:164A;2472-7.
Peschel N, Wright JT, Koster MO et al. Molecular pathway-based classification of ectodermal dysplasias: First five-yearly update. Genes. 2022;13:2327.
ORPHANET. Hypohidrotic ectodermal dysplasia. (Sett 2023 januar). Tilgjengelig fra: URL: https://www.orpha.net
Blüshcke G, Nüsken KD, Schneider H. Prevalence and prevention of severe complications of hypohidrotic ectodermal dysplasia in infancy. Early Hum Dev. 2010;86:397-9.
Kaercher T, Dietz J, Jacobi JD et al. Diagnosis og X-linked hypohidrotic ectodermal dysplasia by meibography and infrared thermography of the eye. Curr Eye Res. 2015;40:884-90.
Hayashi R, Shimomura Y. Update of recent findings in genetic hair disorders. J Dermatol. 2022;49:55-67.
Lexner MO, Bardow A, Hertz JM et al. Anomalies of tooth formation in hypohidrotic ectodermal dysplasia. Int J Paediatr Dent. 2007;17:10-8.
Crossan E, O’Connel AC. Parental perception on oral health-related quality of life and dental features of ectodermal dysplasia and isolated hypodontia in children. BMC Oral Health. 2021;21:510.
Bergendal B, McAllister A, Stecksén-Blicks C. Orofacial dysfunction in ectodermal dysplasias measured using the Nordic Orofacial Yest – Screening protocol. Acta Odontol Scand. 2009;67:377-81.
HELSENORGE. Persontilpasset medisin. (Sett januar 2023). Tilgjengelig fra: URL: https://www.helsenorge.no/persontilpasset-medisin/
Kere J, Srivastava AK Montonen O et al. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in novel transmembrane protein. Nat Genet. 1996;13:409-16.
Gaide O, Schneider P. Permanent correction of an inherited ectodermal dysplasia with recombinant EDA. Nat Med. 2003;9:614-8.
Hermes K, Schneider P, Krieg P et al. Prenatal therapy in developmental disorders: drug targeting via intra-amniotic injection to treat X-linked hypohidrotic ectodermal dysplasia. J Invest Dermatol. 2014;134:2985-7.
Schneider H, Faschingbauer F, Schuepbach-Mallepell S. Prenatal correction of X-linked hypohidrotic ectodermal dysplasia. N Engl J Med. 2018;378:1604-10.
Clinical trials. Intraamniotic administrations of ER004 to male subjects with X-linked hypohidrotic ectodermal dysplasia (EDLIFE). (Sett 2023 januar). Tilgjengelig fra: URL: https://clinicaltrials.gov/ct2/show/NCT04980638?cond=Hypohidrotic+Ectodermal+Dysplasia&draw=3&rank=14
Price JA, Bowden DW, Wright JT et al. Identification of a mutation in DLX3 associated with Trichodento-osseous (TDO) syndrome. Hum Mol Genet. 1998;7:563-9.
Kantaputra PN, Hamada T, Kumchai T et al. Heterozygous mutation in the SAM domain of p63 underlies Rapp-Hodgkin ectodermal dysplasia. J Dent Res. 2003;82:433-7.
Klineberg I, Cameron A, Whittle T et al. Rehabilitation of children with ectodermal dysplasia: Part 1: an international Delphi study. Int J Oral Maxillofac Implants. 2013;28:1090-100.
Hobkirk JA, Nohl F, Bergendal B et al. The management of ectodermal dysplasia and severe hypodontia. International conference statements. J Oral Rehabil. 2006;33:634-7.
Saltnes SS, Geirdal AØ, Saeves R et al. Experiences of daily life and oral rehabilitation in oligodontia – a qualitative study. Acta Odontol Scand. 2019;77:197-204.
Klineberg I, Cameron A, Hobkirk J et al. Rehabilitation of children with ectodermal dysplasia: Part 2: an international consensus meeting. Int J Oral Maxillofac Implants 2013;28:1101-9.
Wang Y, He J, Decker AM. Clinical outcomes of implant therapy in ectodermal dysplasia patients: a systematic review. Int J Oral Maxillofac Surg. 2016;45:1035-43.
Bergendal B, Ekman A, Nilsson P. Implant failure in young children with ectodermal dysplasia: a retrospective evaluation of use and outcome of dental implant treatment in children in Sweden. Int J Oral Maxillofac Implants. 2008;23:520-4.
Chrcanovic BR, Abreu MHNG. Survival and complications of zygomatic implants: a systematic review. Oral Maxillofac Surg. 2013;17:81-93.
Goker F, Grecchi E, Mancini EG et al. Zygomatic implant survival in 9 ectodermal dysplasia patients with 3.5- to 7-year follow-up. Oral Dis. 2020;26:1803-9.
Yin W, Bian Z. Hypodontia, a prospective marker for tumor? Oral Dis. 2016;22:265-73.
English summary
Congenital tooth agenesis and ectodermal dysplasias
Nor Tannlegeforen Tid. 2024; 134: 306-11.
Agenesis of one or a few teeth is common in the population, while congenitally missing six or more teeth (oligodontia) is relatively uncommon. Hypodontia can be an isolated condition or part of a syndrome, of which the various ectodermal dysplasias are among the most common. Hypohidrotic ectodermal dysplasia is the best known, and the best described, form of ectodermal dysplasia and boys with this diagnosis miss an average of 22 permanent teeth. In addition, manifestations are seen in other tissues, such as hair, skin and exocrine glands, and this requires multidisciplinary follow-up and treatment. The dental team has an important and challenging role in both follow-up and treatment of people of all ages with ectodermal dysplasia.
Korrespondanceansvarlig andenforfatter: Hilde Nordgarden, e-post: hilde.nordgarden@tako.no
Akseptert for publisering 21. februar 2023, i Tandlægebladet 2023;127:776-81
Artikkelen er fagfellevurdert
Artikkelen siteres som: Theisen OR, Nordgarden H. Medfødt tandmangel og ektodermale dysplasier. Nor Tannlegeforen Tid. 2024; 134: 306-11.
Emneord: Hypodontia; Oligodontia; Ectodermal dysplasia