
Oral manifestations of systemic disorders – part 1
Headlines
Oral manifestations may be the first sign of a systemic condition or disease
Clinical recognition of oral manifestations related to systemic diseases may be difficult and can delay diagnosis
Oral conditions related to the treatment of systemic diseases are clinically important to recognize
Oral health personnel should be educated to be able to identify and diagnose and manage oral manifestations of systemic diseases and conditions in a timely manner
Oral tissues can be subject to change or damage because of a disorder that predominantly affects other body systems. Such oral manifestations of systemic disorders can be highly variable in both frequency and presentation. Oral manifestations of systemic disorders may present as initial symptoms of an acute or chronic systemic condition and can precede years before systemic symptoms appear and diagnosis of the disease. Additionally, pathological processes in the oral mucosa, jaws and related structures can impact the patient’s overall health. As lifespan increases and medical care becomes ever more complex and effective, it is likely that the numbers of individuals with oral manifestations of systemic disorders will continue to rise. It is important that potential oral manifestations of systemic disorders are managed quickly and appropriately to improve the patient’s quality of life. Oral manifestations may be the first evidence of an underlying systemic disease and may accompany or precede the diagnosis of the disease. The general dentist thus plays an essential role in the detection and referral of patients with possible systemic diseases. When oral findings are accompanied by abdominal pain, diarrhea and intestinal symptoms, this should alert the dentist to quickly refer the patient for further medical management. Dental follow-up in collaboration with the treatment team is important since exacerbation of oral manifestations may indicate increased disease activity.
The oral cavity and orofacial region can exhibit signs and symptoms indicative of a systemic disorder, whether preexisting, newly discovered or during the course of the disease. These manifestations may present as initial symptoms of an acute or chronic systemic condition and can precede years before systemic symptoms appear and diagnosis of the disease. Worsening of an existing disease may cause oral manifestations and symptoms. Additionally, pathological processes in the oral mucosa, jaws and related structures can impact the patient’s overall health directly or indirectly. Furthermore, systemic conditions can impair the function of the orofacial region or affect the patient’s oral health profile.
Part 1 of this article will address the impact of granulomatous diseases and various types of anaemia. In part 2, medication-related osteonecrosis of the jaw, osteoradionecrosis, chronic graft versus host disease and viral infections are discussed. Oral manifestations of dermatological diseases are described in the article Common oral mucosal diseases and lesions and Rare oral mucosal diseases and lesions.
Oral manifestations of systemic conditions and diseases
Granulomatous diseases
Granulomatous diseases belong to a large group of diseases sharing the common histological feature of granuloma formation. A granuloma is a focal aggregation of inflammatory cells, including macrophages, that undergo fusion with other macrophages to form multinucleated giant cells. Of granulomatous diseases, orofacial granulomatosis (OFG), Crohn’s disease (CD), and Melkerson-Rosenthal syndrome (MRS) are of particular importance for dentists.
Orofacial granulomatosis (OFG)
Definition
Orofacial granulomatosis (OFG) is a chronic granulomatous disease affecting the oral mucosa and the orofacial region. OFG may precede the development of Crohn’s disease (CD), appear concomitantly with CD or occur after the diagnosis of CD [1]. The association between OFG and CD has been known for several decades, as the two conditions share both clinical and histopathological features, but the reason is yet unclear. Opinions differ regarding whether OFG should be referred to as a separate entity or as a part of the CD spectrum when it is present with CD [1]. In fact, there has been an ongoing debate as to what constitutes the correct terminology. Some researchers state that OFG, when it appears in association with intestinal CD, should be referred to as oral CD, while others propose that OFG should be considered as a separate entity.
Age and incidence
OFG may occur at any age, but it usually affects children and young adults [1] [2]. It is quite an uncommon disease with a prevalence suggested to be between < 0.1 - 0.8%, and the Celtic population appears to have the highest prevalence [3]. The terminology confusion makes it difficult to compare data from one country to another. Some studies distinguish between OFG and OFG with concomitant CD, while other reports do not make this distinction.
Predisposing factors
Several predisposing factors have been suggested, such as environmental and immunological factors as well as infections and allergic reactions. OFG is a complex condition and most likely multifactorial. The association with CD has led researchers to look at genetic variations in the nucleotide-binding oligomerization domain-containing protein 2 (NOD2) gene, but there is hitherto no supporting data suggesting genetic susceptibility to OFG [3] [4]. It is a very complex condition with many potential causes and contributing factors.
Clinical features
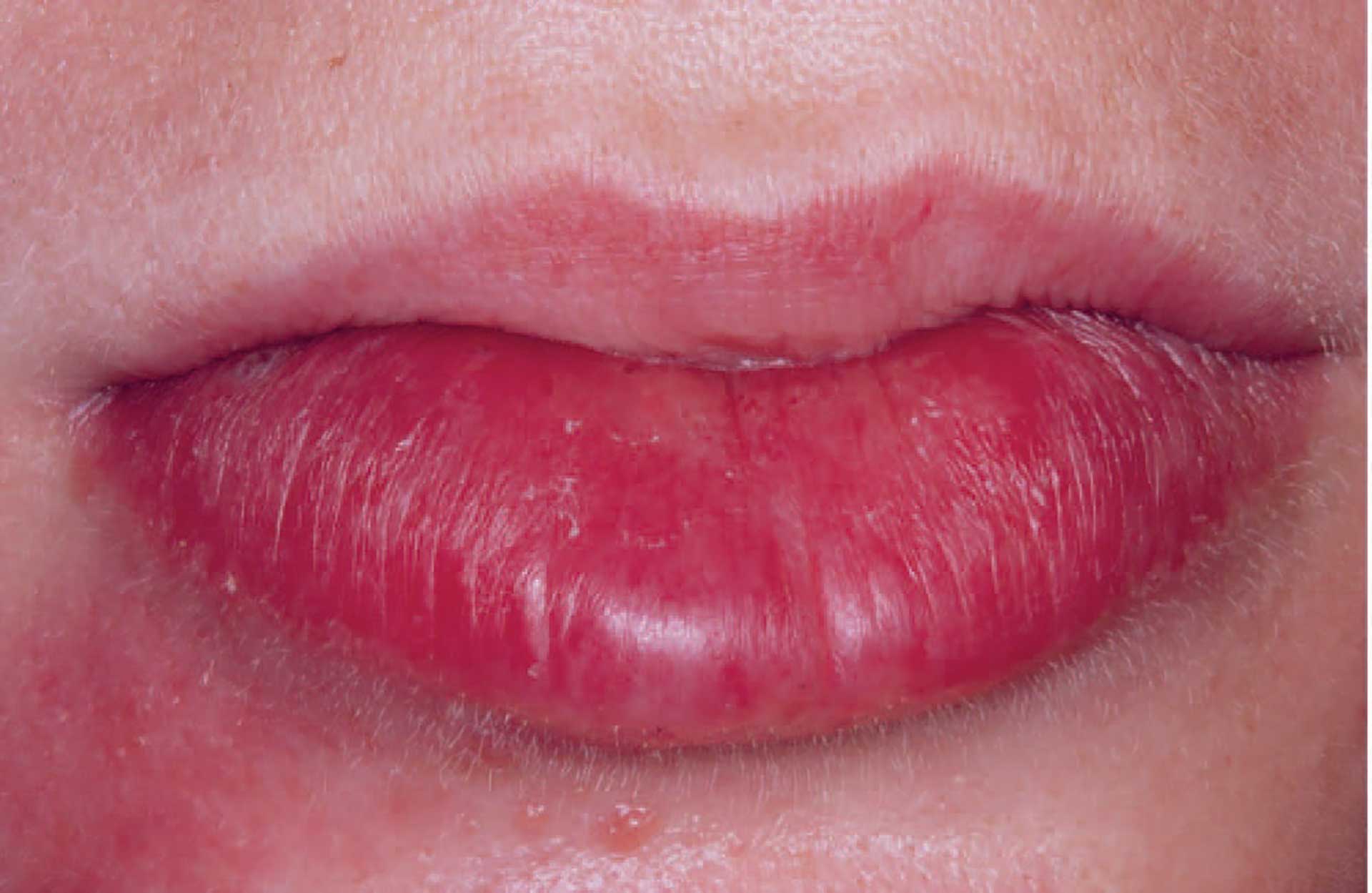
Figure 1. Swelling of the labial mucosa in orofacial granulomatosis.
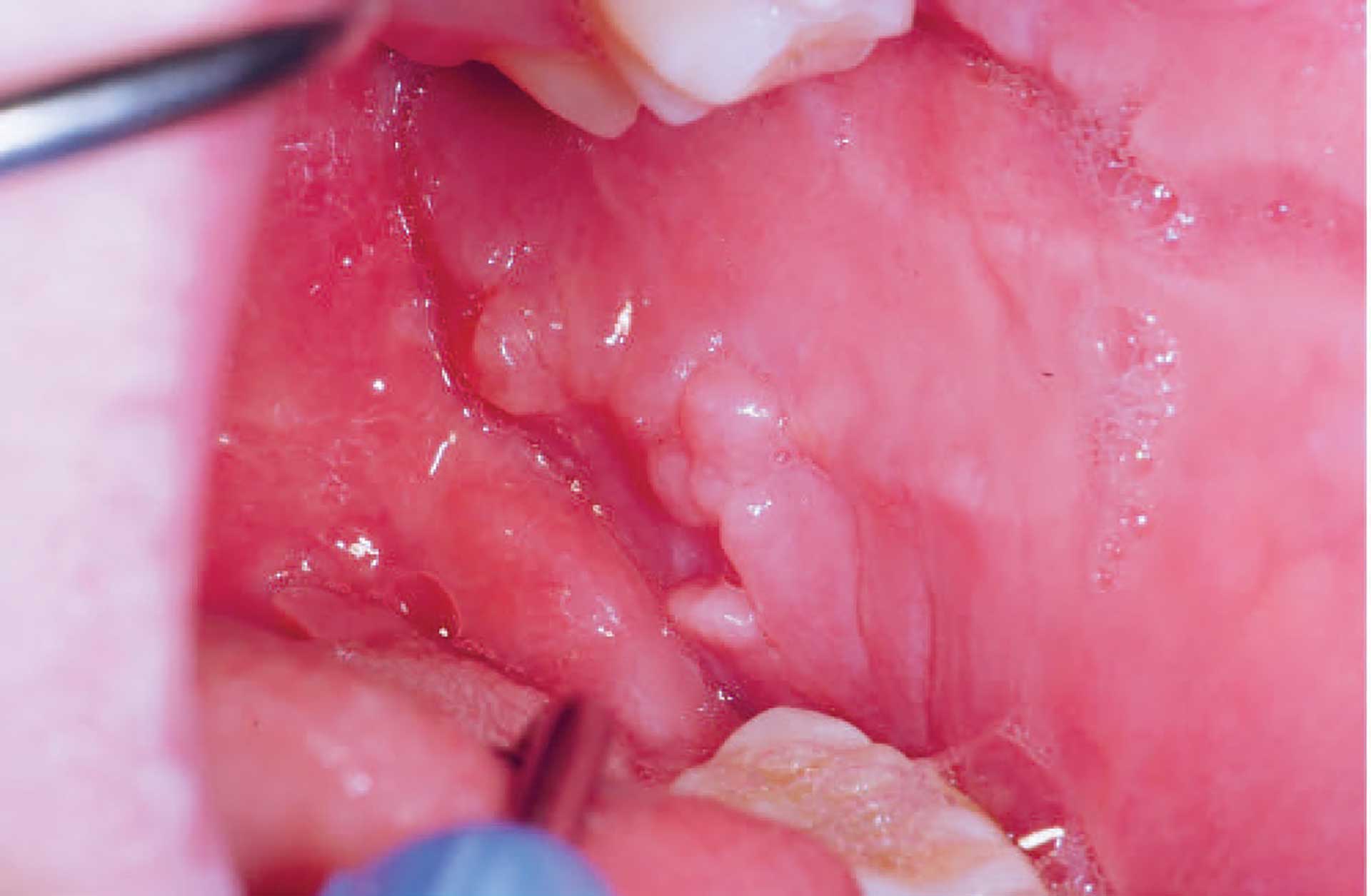
Figure 2. Cobblestone phenomenon in the left buccal mucosa in orofacial granulomatosis.
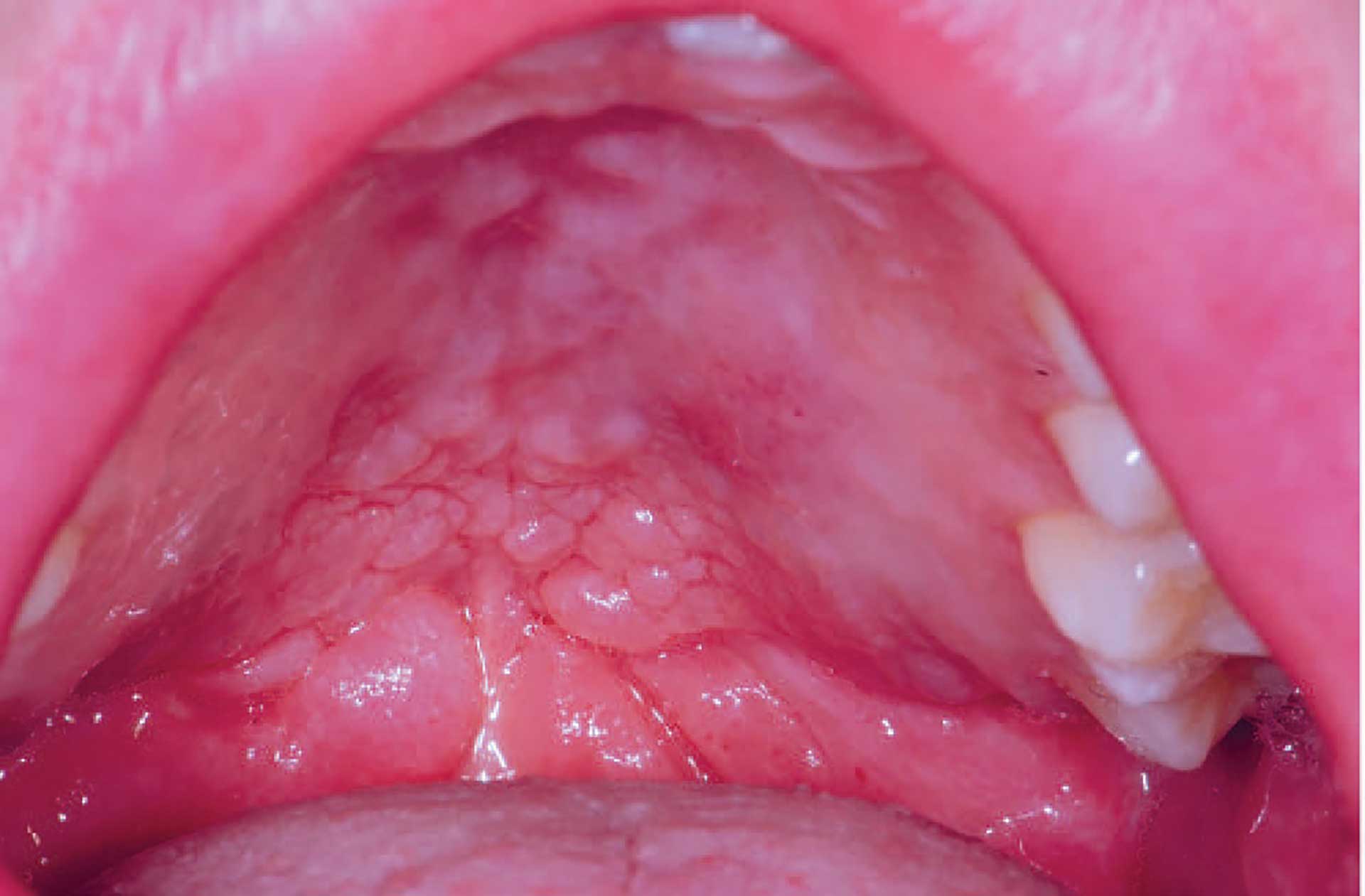
Figure 3. Cobblestone phenomenon in the palate in orofacial granulomatosis.
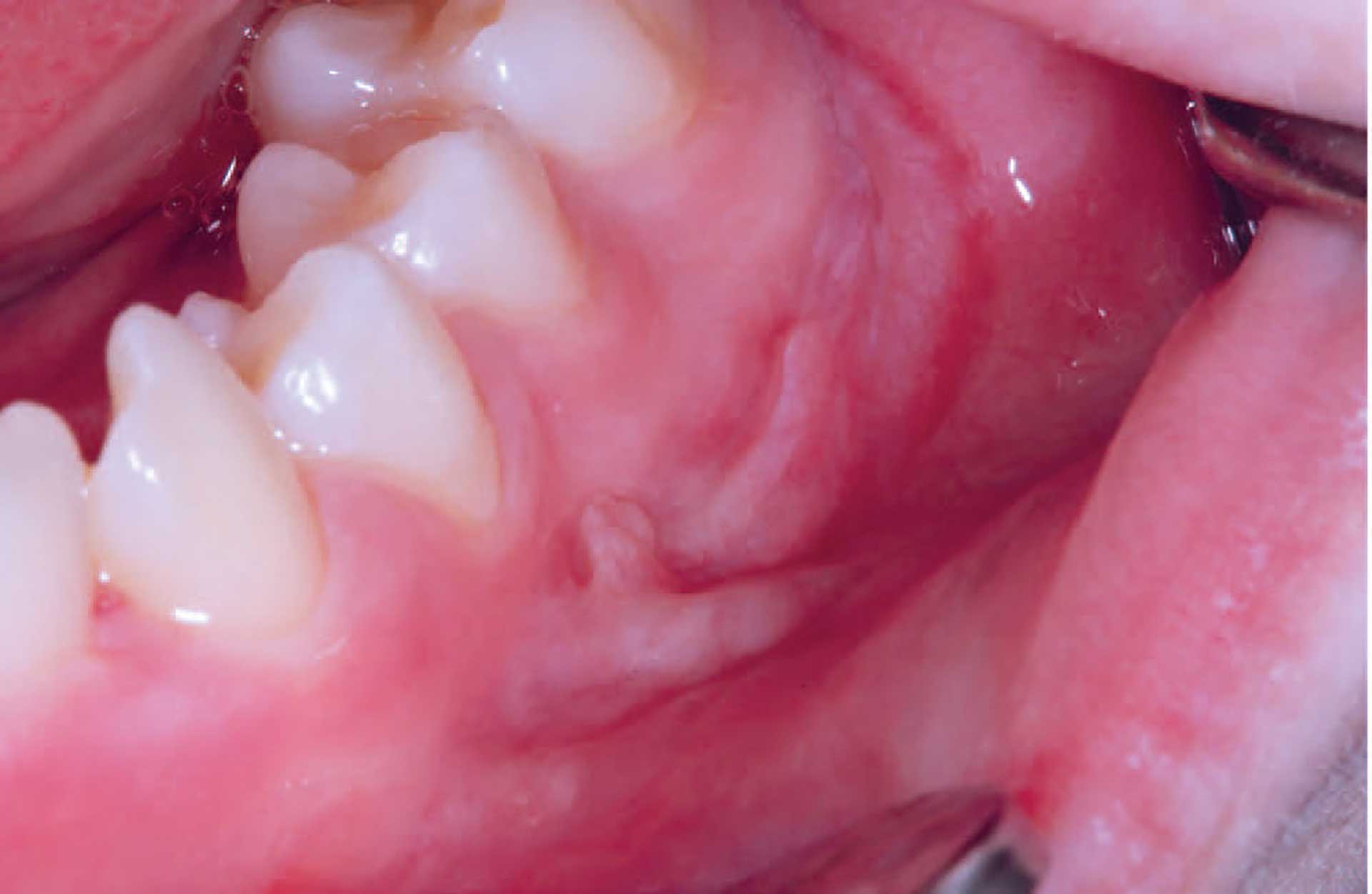
Figure 4. Mucosal tag formation in orofacial granulomatosis.
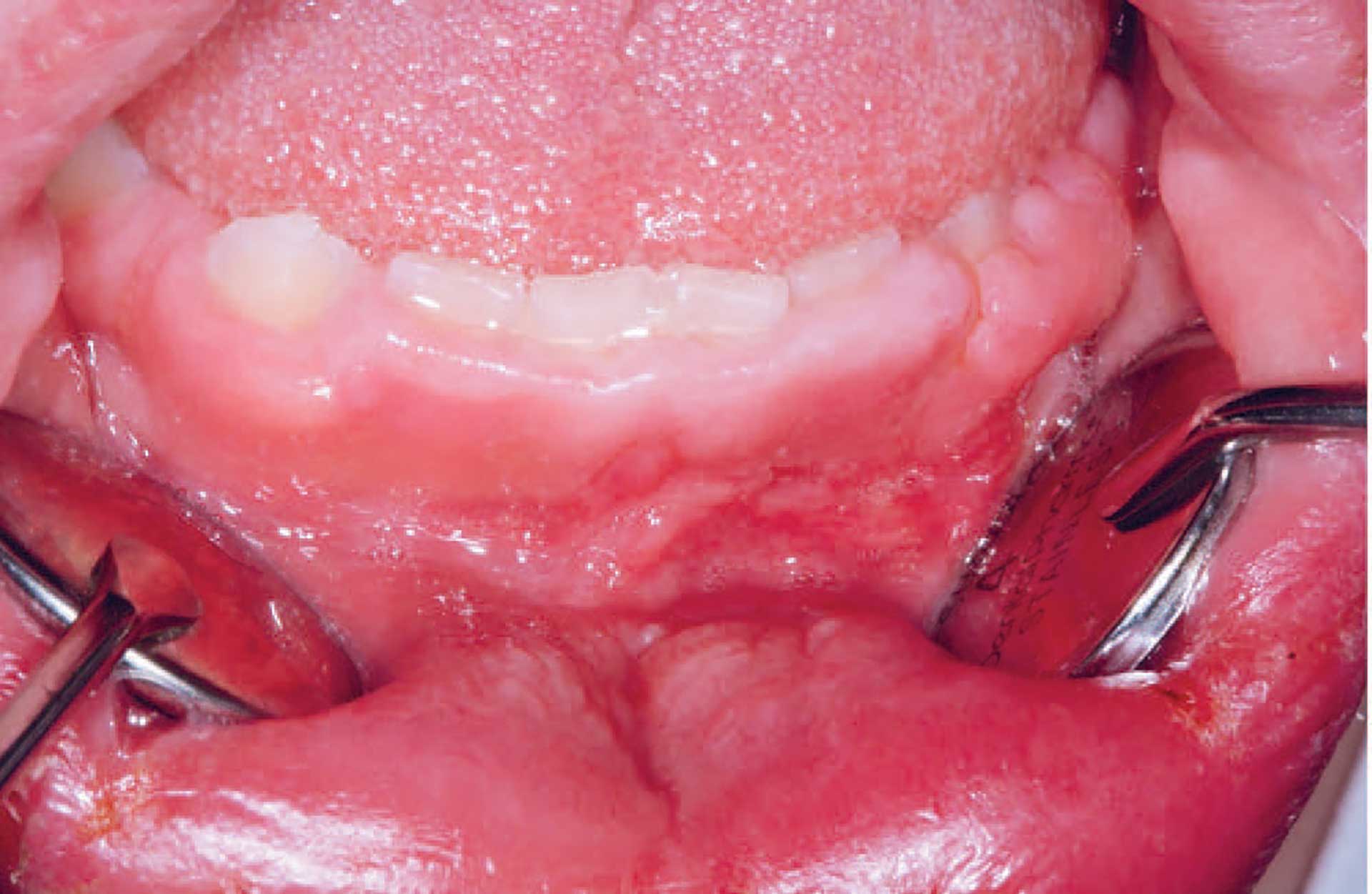
Figure 5. Gingival hyperplasia in orofacial granulomatosis.
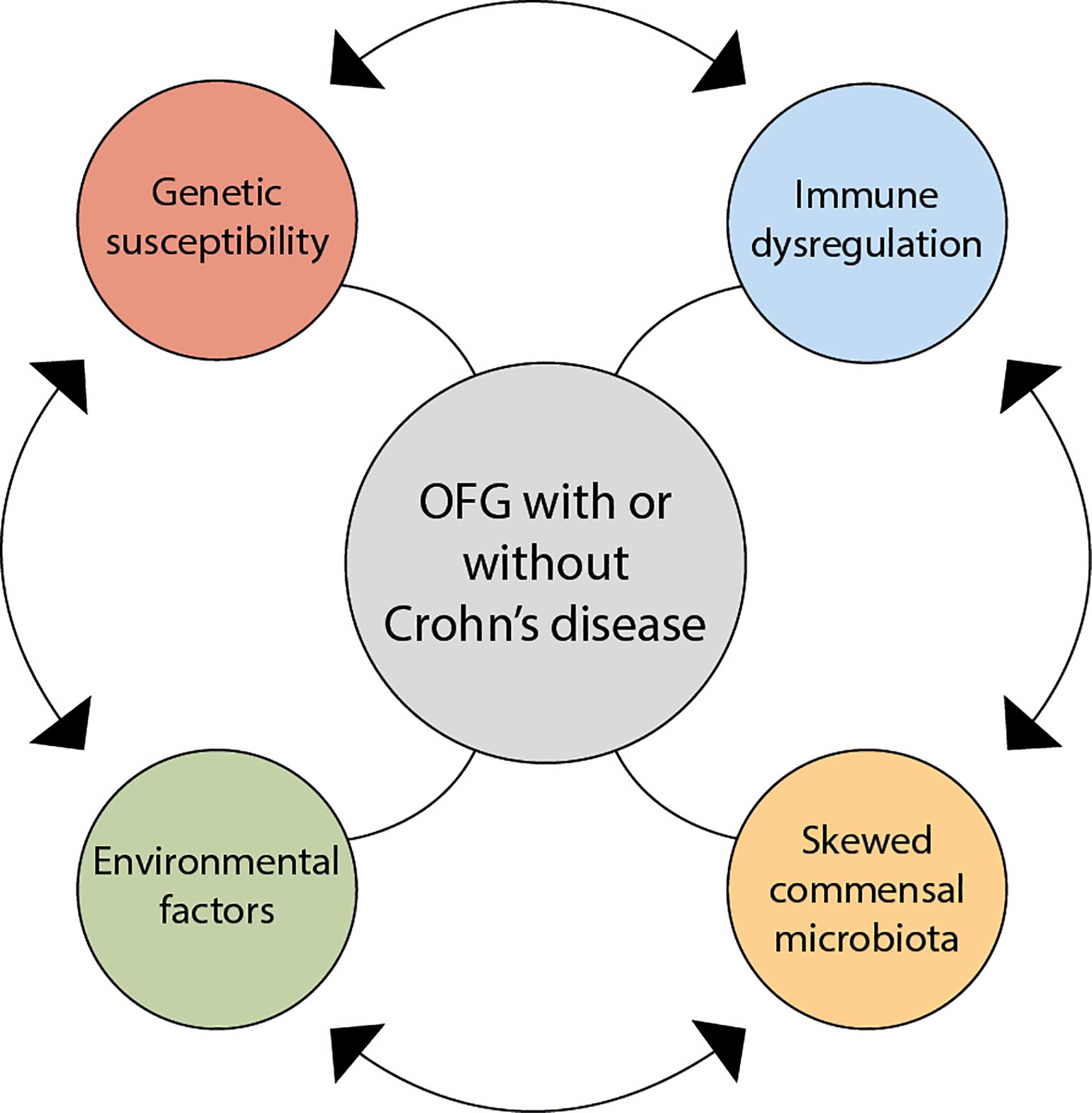
Figure 6. A plausible theory behind the aetiology of orofacial granulomatosis.
The clinical features of OFG vary from one individual to another. The lips, cheeks, gingiva, tongue, palate, and floor of the mouth may be involved. The most common signs are labial and facial swellings, which may develop over several hours to days, thus mimicking angioedema and IgE-mediated allergic reactions. OFG may also cause lesions such as angular cheilitis, the cobblestone phenomenon, ulceration, tag formation, mucosal ridges and hyperplastic and erythematous gingiva [1] [5]. Most often, several of the features occur simultaneously (figure 1-5).
Pathogenesis
In most studies, the four frequently suggested causative elements are environmental influences, genetic susceptibility, immune dysregulation, and a skewed commensal microbiota. Hypersensitivity to food constituents and food additives is suggested in several studies as an aetiological factor, as 60-68% of patients with OFG report food allergies [6] [7]. Genetic susceptibility coincides with OFG and its association with CD, but there is no evidence for genetic susceptibility with respect to OFG [3] [4]. Immune dysregulation could be a result of an abnormal immune response to a trigger antigen. Studies analysing the presence of T cells, B cells and macrophages have suggested that the immune response in OFG is of the T helper 1 type [8] [9]. Microbial dysbiosis may play a role in the development of OFG, although the exact mechanism of this is not clear [10] [11]. It is difficult to envision whether any one of the four elements is more important than the other (figure 6).
Diagnosis
The diagnosis is based on clinical, histological, and immunopathological findings together with anamnestic information. Unfortunately, there is often a delay due to challenges in interpreting the clinical signs and symptoms. The clinical features together with anamnestic information are sufficient to diagnose OFG, as the biopsy does not always show the characteristic granuloma formation. An incisional biopsy should be deep (taken into the connective tissue underneath the epithelium) and performed in an area with swelling and a palpable mass.
Differential diagnostics
Melkerson-Rosenthal syndrome (MRS) is a rare condition that causes a triad of similar orofacial symptoms (see below). Similar to OFG, the disease often begins in childhood or adolescence. Sarcoidosis is a granulomatous disorder typically affecting the lungs, but it can appear in other organs, such as the skin, lymph nodes and mouth. When occurring in oral mucosa, it can cause erythema, ulcerations and noncaseating granulomas. Angioedema is a condition causing orofacial swelling, with lips being the most frequent site. Hypersensitivity or allergic reactions to food, medication or dental material may result in swelling and inflammation in the orofacial region. Both angioedema and hypersensitivity most often have a quick onset. Granulomatosis with polyangiitis (GPA) (formerly called Wegner’s granulomatosis) may also affect the oral mucosa, causing granulomatous inflammation and necrotizing vasculitis of small vessels [12]. GPA is characterized by rhinosinusal signs and lung and kidney involvement. It has a predilection in the upper airways. “Strawberry-like” gingivitis may be the first sign of GPA in the oral cavity. A referral to an internist is recommended. Histopathological findings of granulomas may also be a result of a foreign body reaction to materials such as dental fillings or orthodontic appliances. Tuberculosis is also a chronic granulomatous disease, typically of the lungs, but potentially involving multiple body systems and the oral cavity. A higher prevalence of tuberculosis is found in developing countries or among immunocompromised patients. It may involve soft tissue of the oral cavity and the jawbone, mostly the mandible. Possible routes of infection are sites of recent dental extraction, through mucosal wounds and haematogenous and/or lymphatic spread. Clinically, tuberculosis may resemble periapical lesions, abscesses or ulceration of oral mucosa associated or not with lymphadenopathy.
Treatment
In patients with OFG, it is important to establish whether the oral signs and symptoms are related to gastrointestinal inflammation. Studies show that 54% of patients with OFG and without gastrointestinal symptoms present with macro- and/or microscopic inflammation in biopsies from ileocolonoscopy [1]. Therefore, professionals should consider referrals for gastrointestinal evaluation. It is important to have a multidisciplinary approach regarding the treatment. Dietary modifications and avoidance of triggering foods should be one of the first aspects in the management of OFG. Benzoates and cinnamon have been suggested to trigger or cause exacerbation of the condition, and studies have shown that a benzoate- and cinnamon-free diet has had a good effect on symptoms [6] [13]. Food sensitivity testing is recommended.
Various medications may be used to manage the inflammation associated with OFG, including topical and systemic corticosteroids, immunomodulators, and antibiotics.
Prognosis
OFG is a chronic disease with relapse. The prognosis depends on the severity of the disease and the treatment response. Patients with OFG with concomitant CD have a more severe phenotype of OFG and therefore a worse outcome.
Inflammatory bowel disease
Inflammatory bowel disease (IBD) comprises two main diseases affecting the gastrointestinal tract, i.e., Crohn’s disease (CD) and ulcerative colitis (UC). They are chronic idiopathic progressive disorders affecting the entire gastrointestinal tract [14]. CD can affect any part of the gastrointestinal tract, the most common being the terminal ileum and colon, while UC is seen in the large intestine. Extraintestinal manifestations are present in 5-50% of all IBD cases and include uveitis, spondyloarthropathy and primary sclerosing cholangitis (PSC) [15]. The latter should be diagnosed as early as possible because PSC is associated with an increased malignancy risk of cholangiocarcinoma [16]. Oral manifestations may precede years before systemic symptoms and the diagnosis of IBD. Thus, dental practitioners play an important role in the early detection and treatment of oral lesions.
CD is a long-standing, relapsing disease presenting with segmental and transmural intestinal inflammation. The oral mucosa may be involved. The presence of granulomas in the intestinal mucosa is one of the hallmarks of the disease. The onset of disease during childhood may result in impaired growth when untreated [17] [18]. When oral findings appear in association with intestinal CD, some refer to them as oral CD. In other cases, the findings are regarded as OFG and considered a separate entity [1].
UC is also a relapsing and chronic inflammatory bowel disease (IBD) characterized by mucosal inflammation that starts distally and can extend proximally to involve the whole colon [19].
Age and incidence
The incidence of IBD is increasing worldwide, especially in newly industrialized countries, with the incidence of CD being between 0.4 and 22.8 per 100 000 person-years and UC between 2.4 and 44.0 per 100 000 person-years [14] [15]. Sweden, together with Canada and Scotland, has the highest incidence rates in the world.
The prevalence of oral manifestations in IBD is reported to range from 0.7% to 37% in adults and 7% to 23% in children [20] [21] [22] [23]. The variability in the prevalence may be due to different study designs and due to the number and type of study populations.
CD may appear at any age, although most individuals are diagnosed before 30 years of age. There is also a small peak in incidence between 70 and 80 years of age. Early childhood infections have been put forward as having a protective effect on CD [24]. These childhood infections are supposed to create a balance between proinflammatory and tolerance-inducing mechanisms that in turn will protect against inflammatory responses to antigen stimuli. Several studies show that developed countries with higher life quality standards have a higher rate of CD incidence than developing countries.
UC may appear at any age. A bimodal age distribution has been reported with a peak in the second or third decades and a peak between 50 and 80 years, with the highest incidence of UC among people aged 20-29 years [25].
Clinical features
The clinical features of IBD vary from one individual to another, but most patients experience abdominal pain together with diarrhea as well as weight loss. It is not uncommon that patients with IBD experience fever and fatigue. Oral manifestations of IBD can be specific or nonspecific due to malnutrition and drugs. Aphthous-like ulcers, labial swelling, angular cheilitis, mucosal tags and cobblestone-like mucosal lesions are more indicative of CD. Oral mucosal ulcers are associated with the active phase of CD, resembling ulcers seen in the gastrointestinal tract. However, oral ulcers may persist and stay chronic on the same site without healing.
The oral manifestations of UC include specific and nonspecific features, with the former including pyostomatitis vegetans (PV), characterized by pustules in the gingiva, and the latter including recurrent aphthous-like ulcers, atrophic glossitis, oral mucosal burning sensation, angular cheilitis, dry mouth, taste disturbances, halitosis and periodontitis [25] [26]. The oral manifestations may be asymptomatic and occur prior to or parallel with UC activity. A higher frequency of dental caries and periodontal disease has been reported in patients with IBD [23] [27].
Pathogenesis
The etiopathology of IBD is still unknown but is considered multifactorial with a complex interplay between genetic susceptibility, altered gut microbiota and environmental factors, leading to dysregulated immune responses [28]. However, it is difficult to envision whether any one of them is more important than the other.
Genetic susceptibility is a well-known risk factor for the development of CD. A Swedish study from 2013 showed that 14% of patients with CD had a relative with IBD. Genome-wide association studies have identified more than 200 alleles associated with IBD, of which 37 are specific for Crohn’s disease [29] [30] [31].
Predisposing factors
As with OFG, several predisposing factors have been suggested for both CD and UC, such as environmental factors, genetic susceptibility, immune dysregulation and imbalanced commensal microbiota. Delayed type hypersensitivity to food constituents and food additives, improved hygiene, decreased early childhood infections, and tobacco usage are some environmental factors that could increase the risk of developing CD [32] [33]. The theory of familial IBD comprising first-degree relatives with the condition was accepted at the end of the 1950s, but it was not until 2001 that the association between NOD2 gene variations and CD was demonstrated [3] [4]. Later, reports showed that NOD2 variations are associated with a more complicated CD. UC is suggested to be a modified T-helper 2 disease in which the innate immune system initiates inflammatory events with the adaptive immune system [25].
There are several indications of how the microbiota seems to affect the development of IBD, but no specific organism has been suggested as the key pathogenic microbe. Instead, there seems to be dysbiosis, i.e., microbial imbalance, affecting host-microbe interactions [34].
This dysbiosis seems to be caused by the impaired ability of the gut mucosa to kill bacteria [34]. Early childhood infections are supposed to create a balance between proinflammatory and tolerance-inducing mechanisms that in turn protect against inflammatory responses to antigen stimuli. Oral tolerance is important for the avoidance of T cells hyperresponsive to pathogenic microbiota as well as commensal microbiota. Oral tolerance actively suppresses immune responses to nondangerous dietary antigens or commensal bacteria, which starts to develop during infancy [35].
Improved hygiene and use of antibiotics in developed countries has been a frequently suggested risk factor in the pathogenesis of IBD [30] [31]. This risk factor is supported by the increased incidence rates seen in countries changing status from a developing to a developed country.
Cigarette smoking is the best studied environmental factor and is associated with a twofold increased risk of CD. Interestingly, smokers may be protected from the onset of UC, but formal smoking is known as a risk factor for the development of UC [36]. Other risk factors include antibiotic exposure in childhood and reduction in dietary fibre.
Diagnosis
The diagnosis of IBD is based on a combination of several tests, including clinical, biochemical, stool-test (calprotectin), endoscopic, cross-sectional imaging, and histological evaluations. There is no single diagnostic test. Dentists may contribute to the diagnosis when oral manifestations are suspected to be associated with systemic diseases.
The histopathological characteristics of CD include granuloma formation and focal crypt architectural abnormalities, together with focal or patchy chronic inflammation with the presence of lymphocytes and plasma cells, along with mucin preservation at active sites. To date, there is no consensus on how many histopathological features need to be present in the biopsy to make the final diagnosis. However, the presence of epithelioid granulomas and an additional feature is sufficient to confirm the CD diagnosis [37]. There are no specific diagnostic histological characteristics of UC, but basal plasmocytosis, crypt atrophy/distortion and villous surface irregularities are suggested signs of UC. The presence of granulomas is more suggestive of CD [19].
Differential diagnostics
Irritable bowel syndrome (IBS) is a gastrointestinal condition with features such as abdominal pain, bloating, and deviant bowel movements. Infectious colitis is caused by bacterial or viral infections, and the symptoms may mimic the symptoms of CD. Diverticulitis may also cause inflammation or infection of small pouches in the colon wall, resulting in abdominal pain. Celiac disease is an autoimmune disease due to intolerance of gluten and with clinical features of abdominal pain, diarrhea, and malabsorption.
Treatment
IBD is treated with anti-inflammatory drugs, immunosuppressants (i.e., corticosteroids, 5-aminosalicylates, azathioprine and methotrexate) and biologics (i.e., infliximab (anti-TNF-α), vedolizumab and ustekinumab), which has changed the therapeutic landscape. Treatment with antibiotics and dietary supplements, including vitamins and minerals, may also be needed. Nutritional therapy is a key component in its management, and many patients will eventually require surgery, although it is never a cure [38]. In patients with oral mucosal involvement, treatment with topical steroids is often needed.
Prognosis
The prognosis of IBD is dependent on several factors, such as disease severity, compliance, lifestyle, and diet. It is important to consider quality of life and not just the disease outcome when looking at prognosis and complications. Extraintestinal findings such as oral manifestations have been connected to a more severe course of IBD [36].
Melkersson-Rosenthal syndrome
Definition
Melkersson-Rosenthal syndrome (MRS) is a neuromucocutaneous disorder. It may appear at different periods as monosymptomatic or oligosymptomatic. The most common monosymptomatic variant is Miescher’s cheilitis with granulomatous enlargement of the lips.
Age and incidence
The mean age of onset of MRS seems to vary between 14 and 39 years of age, but the symptoms often start in the 2nd decade of life. However, it may occur at any age. The incidence of MRS is suggested to be between 0.2 and 80 in 100,000 per year. There is no gender difference in the prevalence of MRS and no racial propensity. The varied incidence reports may be because of underestimated incidence [39] [40].
Predisposing factors
Several associating factors have been postulated as a risk of developing MRS. Genetic propensity to MRS by increased expression of certain HLA types has been suggested. Infectious and immunologic factors have also been discussed as well as hypersensitivity reactions, although to date, nothing has been proven [41] [42].
Clinical features
There is a triad of recurrent symptoms in MRS, including orofacial swelling, relapsing facial paralysis, and fissured tongue (lingua plicata). The most common symptom is orofacial swelling, and the affected area is most often the upper lip. However, other areas, such as cheeks, chin and periorbital tissues, may also be affected. The swelling may be persistent, but most often it is relapsing. Regarding facial palsy, it is usually recurrent as well, although occasionally it can be persistent. Usually, facial palsy is unilateral; however, approximately two-thirds of patients who initially present with unilateral symptoms develop contralateral facial palsy. The fissured tongue is most often asymptomatic. The histopathological findings are noncaseating epithelioid granulomas, perivascular lymphocytic infiltration, and edema [43].
Pathogenesis
The etiology of MRS is unknown, but varying theories have been put forward regarding the underlying mechanisms, including food hypersensitivity, reactions to dental materials, bacterial and viral infections, immune dysregulation, association with IBD, and genetic predisposition [44]. To date, no specific food allergens or dental materials are known.
Diagnosis
Recurring or persisting orofacial oedema concomitant with the presence of at least one of the findings of facial palsy or lingua plicata is sufficient to make an MRS diagnosis. Only 8-25% of the cases show the complete triad, with orofacial oedema being the most common symptom. Histopathological analysis is of diagnostic value even though clinical symptoms may be sufficient for diagnosis. The disorder is often underdiagnosed, and a mean diagnostic delay is estimated to be as high as nine years.
Differential diagnostics
Both OFG and CD need to be ruled out, as MRS may mimic these conditions. With orofacial swelling being the most common MRS symptom, it is easy to mistake the disorder for OFG or CD.
Treatment
Symptomatic treatment may include nonsteroidal anti-inflammatory drugs (NSAIDs) and corticosteroids to reduce swelling. Local or intralesional corticosteroids may improve the clinical and histological picture, although it is often temporary. Antibiotics such as roxithromycin and minocycline have also proven to have a positive effect on symptoms. As with CD, immunosuppressants may also have a positive effect on MRS.
Prognosis
MRS may be relapsing or persistent, but good oral hygiene and avoidance of triggering factors together with appropriate medication will give the best conditions for a good prognosis.
Iron-deficiency anaemia and anaemia of chronic diseases
Definition
The World Health Organization (WHO) defines anaemia as a haemoglobin level less than 13 g/dL in men, less than 12 g/dL in women (pregnancy < 11 g/dL) and below <11 g/dL in children [45]. Anemia is a symptom, and the cause of anemia should be investigated.
Iron is an important component of the haemoglobin molecule. Iron-deficiency anaemia is the most common type of anaemia worldwide. The second most common type is anaemia of a chronic disease. It may occur in autoimmune diseases (i.e., rheumatoid arthritis), cancer (hematological and solid tumors), infections and chronic renal diseases. It is driven by activation of the immune system by autoantigens, tumour antigens or microbial molecules, leading to the release of cytokines, which suppress erythropoiesis, decrease erythropoietin, shorten the half-life of erythrocytes, and elevate serum levels of hepcidin and hypoferraemia [46] [47].
Predisposing factors/etiology
The main cause of iron deficiency anaemia is acute or chronic blood loss, i.e., excessive menstrual blood loss, gastrointestinal bleeding, inflammatory bowel disease (IBD), gastrointestinal cancer and medication-induced gastric ulcers (due to NSAIDs, acetylsalicylic acid, anticoagulants or corticosteroids). Additional causes include an insufficient dietary intake of iron, liver diseases, alcohol abuse, an increased demand for erythrocyte production during childhood growth spurts and during pregnancy, a reduced absorption of iron (i.e., in patients with total gastrectomy or celiac disease) and haemolysis [46] [47].
Clinical features
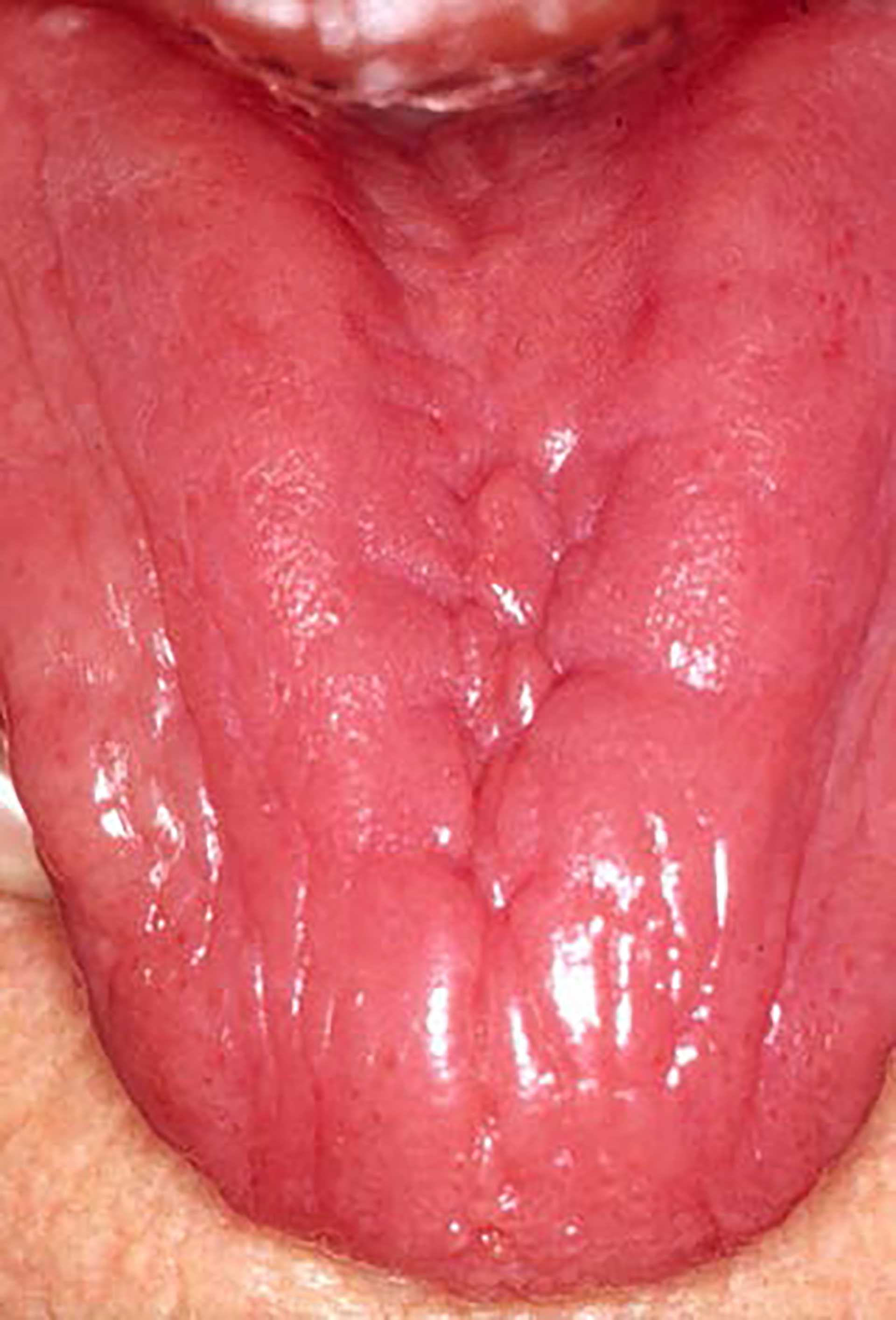
Figure 7. The dorsum of the tongue with atrophy of the tongue papilla in a patient suffering from anaemia.
The spectrum of clinical manifestations is broad and depends on the severity of the anaemia. Fatigue, dyspnoea, weakness, dizziness, headache, heart palpitations, pica, pallor of the skin, and koilonychia are common symptoms and signs. Oral symptoms and signs often include atrophic glossitis, erythematous macules or diffuse mucosal erythema, angular cheilitis, candidiasis, glossodynia, soreness and burning sensation of the oral mucosa, sensation of oral dryness and reduced taste sensitivity (figure 7). Plummer Vinson syndrome is a type of iron-deficiency anaemia that is characterized by dysphagia, angular cheilitis and atrophic glossitis. It is classified as a potentially malignant oral mucosal condition that predisposes patients to the development of squamous cell carcinoma.
Diagnosis and treatment
The clinical diagnosis of anaemia should be confirmed by laboratory tests. The cause of anaemia should always be examined to rule out any underlying disease. In cases of iron deficiency, the anaemia is usually microcytic, whereas it is normocytic and normochromic in anaemia of chronic disease (table 1). Iron-deficiency anaemia and anaemia of chronic disease may occur simultaneously. The treatment of iron-deficiency anaemia as well as its oral manifestations includes treating the underlying cause, such as gastrointestinal bleeding and oral iron supplementation.
Iron-deficiency anaemia |
Anaemia of chronic disease |
Vitamin B12 deficiency |
Folate deficiency |
|
|---|---|---|---|---|
Haemoglobin |
< 13 g/dL in men < 12 g/dL in women |
Low |
Low |
Low |
Ery-RDW (anisocytosis) |
Low <15% |
Low or normal |
High |
High |
MCV |
Low <80 fL |
Normal or low |
High >100 fL |
High >100 fL |
MCH |
Low <27 pg |
Normal |
High |
High |
Transferrin |
High |
Normal |
Normal |
Normal |
Iron level |
Low |
Low |
Normal or high |
Normal or high |
Ferritin |
Low < 30 ng/mL |
High >100 ng/mL |
Normal |
Normal |
Cobalamin |
Normal* |
Normal |
Low <200 pg/mL |
Normal |
Folic acid |
Normal* |
Normal |
Normal or low |
Low |
Ery-RDW, red cell distribution width; MCV, mean corpuscular volume; MCH, mean corpuscular haemoglobin; *Low if general malabsorption
Vitamin B12 deficiency and folate deficiency
Predisposing factors/etiology
The most common causes of megaloblastic anaemia are vitamin B12 (cobalamin) and vitamin B9 (folate) deficiencies. Both vitamins play a crucial role in the normal maturation of cells since they are necessary for DNA synthesis. Vitamin B12 deficiency may occur due to insufficient dietary intake (i.e., a strict vegan diet, malnutrition), pernicious anemia, gastric resection and intestinal malabsorption (i.e., due to IBD). Pernicious anemia is an autoimmune disease in which both anti-gastric parietal cell and anti-intrinsic factor antibodies can be found. Atrophic gastritis, long-term treatment with proton pump inhibitors, H2-receptor antagonists or metformin, or Helicobacter pylori infection may be associated with impaired absorption of cobalamin due to insufficient pepsin or gastric secretion and inadequate proteolytic digestion [48] [49]. Folate deficiency may occur owing to diminished intake (due to alcohol abuse or malnutrition), increased consumption (due to haemolysis or pregnancy) or malabsorption (due to gastric resection, IBD or medications including cholestyramine or metformin). Medications that impair DNA synthesis include folic acid analogs (i.e., methotrexate, trimethoprim-sulfamethoxazole), nucleic acid analogs (5-fluorouracil, zidovudine), hydroxyurea, pentamidine, phenytoin, pyrimethamine, sulfasalazine, and triamterene. Megaloblastic changes are believed to be caused by impaired DNA synthesis leading to macrocytic erythrocytes, abnormalities in leukocytes and platelets and epithelial changes, especially of epithelial cells lining the oral and gastrointestinal mucosa [48].
Clinical features
Vitamin B12 deficiency may cause a wide range of haematological, gastrointestinal, psychiatric and neurological manifestations. Megaloblastic anaemia is an early and common haematological sign. Peripheral neuropathy, neuropsychiatric disorders and optic nerve atrophy are the most common neurologic manifestations [48]. Clinically, megaloblastic anemia progresses slowly, and symptoms include fatigue, weakness, dyspnea and neurologic abnormalities. Oral signs and symptoms include glossitis, angular cheilitis, recurrent oral ulcers, candidiasis, diffuse erythematous mucosa or pale oral mucosa. [49] [50].
Diagnosis
The diagnosis of vitamin B12 and folate deficiencies includes a thorough history and clinical examination and laboratory tests (Table 1). Concentrations of homocysteine and methylmalonic acid should also be determined. Examination of the underlying cause of vitamin B12 deficiency and folate deficiency should always be carried out. Early diagnosis is crucial for initiating parenteral replacement therapy with cobalamin as early as possible to avoid irreversible neurological damage.
Treatment
The treatment for vitamin B12 or folate deficiency anaemia depends on the underlying cause. Pernicious anemia usually requires parenteral replacement therapy and long-life follow-up of the patients. If the deficiencies are diet-related, parenteral therapy may be necessary initially, followed by peroral treatment until the vitamin levels are restored. Medication-induced B12-vitamin deficiency can be treated with supplement therapy and seldom requires discontinuation of the medicine, e.g., metformin.
Conclusion
Gastrointestinal disorders and/or anaemia may present with oral signs and symptoms. Careful history taking and clinical examination may facilitate determining the underlying etiology of oral manifestations and allow for earlier intervention of the underlying disease. This will decrease patient suffering and reduce permanent damage possibly caused by the disease. Dentists should consider systemic diseases and conditions causing oral manifestations in their differential diagnostics since they play an important role in the early detection and diagnosis of gastrointestinal diseases. Dentists must collaborate with the medical treatment team in the follow-up of patients since oral manifestations and symptoms may mirror systemic disease activity.
References
Sanderson J, Nunes C, Escudier M, Barnard K, Shirlaw P, Odell E, et al. Oro-facial granulomatosis: Crohn’s disease or a new inflammatory bowel disease? Inflamm Bowel Dis. 2005; 11(9):840-6.
Wiesenfeld D, Ferguson MM, Mitchell DN, MacDonald DG, Scully C, Cochran K, et al. Oro-facial granulomatosis--a clinical and pathological analysis. Q J Med. 1985; 54(213):101-13.
Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001; 411(6837):599-603.
Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001 ;411(6837):603-6.
McCartan BE, Healy CM, McCreary CE, Flint SR, Rogers S, Toner ME. Characteristics of patients with orofacial granulomatosis. Oral Dis. 2011; 17(7):696-704.
Campbell H, Escudier MP, Brostoff J, Patel P, Milligan P, Challacombe SJ, et al. Dietary intervention for oral allergy syndrome as a treatment in orofacial granulomatosis: a new approach? J Oral Pathol Med. 2013; 42(7):517-22.
Patel P, Brostoff J, Campbell H, Goel RM, Taylor K, Ray S, et al. Clinical evidence for allergy in orofacial granulomatosis and inflammatory bowel disease. Clin Transl Allergy. 2013; 3(1):26.
Freysdottir J, Zhang S, Tilakaratne WM, Fortune F. Oral biopsies from patients with orofacial granulomatosis with histology resembling Crohn’s disease have a prominent Th1 environment. Inflamm Bowel Dis. 2007; 13(4):439-45.
Patel P, Barone F, Nunes C, Boursier L, Odell E, Escudier M, et al. Subepithelial dendritic B cells in orofacial granulomatosis. Inflamm Bowel Dis. 2010; 16(6):1051-60.
Savage NW, Barnard K, Shirlaw PJ, Rahman D, Mistry M, Escudier MP, et al. Serum and salivary IgA antibody responses to Saccharomyces cerevisiae, Candida albicans and Streptococcus mutans in orofacial granulomatosis and Crohn’s disease. Clin Exp Immunol. 2004; 135(3):483-9.
Gibson J, Wray D, Bagg J. Oral staphylococcal mucositis: A new clinical entity in orofacial granulomatosis and Crohn’s disease. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000; 89(2):171-6.
Ruokonen H, Helve T, Arola J, Hietanen J, Lindqvist C, Hagstrom J. “Strawberry like” gingivitis being the first sign of Wegener’s granulomatosis. Eur J Intern Med. 2009; 20(6):651-3.
Campbell HE, Escudier MP, Patel P, Challacombe SJ, Sanderson JD, Lomer MC. Review article: cinnamon- and benzoate-free diet as a primary treatment for orofacial granulomatosis. Aliment Pharmacol Ther. 2011; 34(7):687-701.
Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017 Dec 23;390(10114):2769-2778. Erratum in: Lancet. 2020; 3;396(10256): e56.
Zhao M, Gönczi L, Lakatos PL, Burisch J. The Burden of Inflammatory Bowel Disease in Europe in 2020. J Crohns Colitis. 2021; 25;15(9):1573-1587.
Dyson JK, Beuers U, Jones DEJ, Lohse AW, Hudson M. Primary sclerosing cholangitis. Lancet 2018; 23;391(10139):2547-2559.
Logan RF. Inflammatory bowel disease incidence: up, down or unchanged? Gut. 1998; 42(3):309-11.
Economou M, Pappas G. New global map of Crohn’s disease: Genetic, environmental, and socioeconomic correlations. Inflamm Bowel Dis. 2008; 14(5):709-20.
Segal JP. LeBlanc JF, Hart AL. Ulcerative colitis: an update. Clinical Medicine 2021;21(2):135-9.
Zippi M, Corrado C, Pica R, Avallone EV, Cassieri C, De Nitto D, et al. Extraintestinal manifestations in a large series of Italian inflammatory bowel disease patients. World. J. Gastroenterol. 2014; 20:17463-67.
Oviedo C, Yañez M, Pennacchiotti V. Frequency of oral manifestation in patients with inflammatory bowel disease in Chile. Int. J. Odontostomat. 2017; 11:267-71.
Greuter T, Bertoldo F, Rechner R, Straumann A, Biedermann L, Zeitz J, et al. Extraintestinal manifestations of pediatric inflammatory bowel disease: Prevalence, presentation and anti-TNF treatment. J. Pediatric. Gastroenterol. 2017; 65:200-6.
Koutsochristou V, Zellos A, Dimakou K, Panayotou I, Siahanidou S, Roma-Giannikou E, et al.A. Dental Caries and Periodontal Disease in Children and Adolescents with Inflammatory Bowel Disease: A Case-Control Study. Inflamm. Bowel. Dis. 2015; 21:1839-46.
Amre DK, Lambrette P, Law L, Krupoves A, Chotard V, Costea F, et al. Investigating the hygiene hypothesis as a risk factor in pediatric onset Crohn’s disease: a case-control study. Am J Gastroenterol. 2006; 101(5):1005-11.
Li C, Wu Y, Xie Y, Zhang Y, Jiang S, Wang J et al.Oral manifestations serve as potential signs of ulcerative colitis: A review. Front Immunol. 2022; 29(13):1013900.
Lauritano D, Boccalari E, Di Stasio D, Della Vella F, Carinci F, Lucchese A, et al. Prevalence of Oral Lesions and Correlation with Intestinal Symptoms of Inflammatory Bowel Disease: A Systematic Review. Diagnostics 2019; 15;9 (3):77.
Grössner-Schreiber B, Fetter T, Hedderich J, Kocher T, Schreiber S, Jepsen S. Prevalence of dental caries and periodontal disease in patients with inflammatory bowel disease: A case-control study. J. Clin. Periodontol. 2006; 33:478-84.
Torres J, Mehandru S, Colombel JF, Peyrin-Biroulet L. Crohn’s disease. Lancet. 2017; 29;389(10080):1741-1755.
Peyrin-Biroulet L, Loftus EV, Colombel JF, Sandborn WJ. The natural history of adult Crohn’s disease in population-base cohorts. Am J Gastroenterol 2010; 105:289-97.
Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY et al, Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012; 491:119-24.
Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 2015; 47:979-86.
Economou M, Pappas G. New global map of Crohn’s disease: Genetic, environmental, and socioeconomic correlations. Inflamm Bowel Dis. 2008; 14(5):709-20.
Carbonnel F, Jantchou P, Monnet E, Cosnes J. Environmental risk factors in Crohn’s disease and ulcerative colitis: an update. Gastroenterol Clin Biol. 2009; 33 Suppl 3:S145-57.
Ott SJ, Musfeldt M, Wenderoth DF, Hampe J, Brant O, Folsch UR, et al. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004; 53(5):685-93.
Hooper LV, Midtvedt T, Gordon JI. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr. 2002; 22:283-307.
Gerhard Rogler, Abha Singh, Arthur Kavanaugh, David T. Rubin, Extraintestinal Manifestations of Inflammatory Bowel Disease: Current Concepts, Treatment, and Implications for Disease Management, Gastroenterology, Volume 161, Issue 4, 2021, P. 1118-32, ISSN 0016-5085.
Tanaka M, Saito H, Fukuda S, Sasaki Y, Munakata A, Kudo H. Simple mucosal biopsy criteria differentiating among Crohn disease, ulcerative colitis, and other forms of colitis: measurement of validity. Scand J Gastroenterol 2000; 35:281–6.
Ruemmele FM, Veres G, Kolho KL, Griffiths A, Levine A, Escher JC, et al. European Crohn’s and Colitis Organisation; European Society of Pediatric Gastroenterology, Hepatology and Nutrition. Consensus guidelines of ECCO/ESPGHAN on the medical management of pediatric Crohn’s disease. J Crohns Colitis. 2014; 8(10):1179-207.
Elias MK, Mateen FJ, Weiler CR. The Melkersson-Rosenthal syndrome: a retrospective study of biopsied cases. J Neurol. 2013; 260:138-43.
Feng S, Yin J, Li J, Song Z, Zhao G. Melkersson-Rosenthal syndrome: a retrospective study of 44 patients. Acta Otolaryngol. 2014; 134:977-81.
Lin T-Y, Chiang C-H, Cheng P-S. Melkersson-Rosenthal syndrome. J Formos Med Assoc. 2016; 115:583-4.
Gavioli CFB, Nico MMS, Panajotopoulos N, Rodrigues H, Rosales CB, Valente NYS, et al. A case- control study of HLA alleles in Brazilian patients with Melkersson- Rosenthal syndrome. Eur J Med Genet. 2020; 63(7):103879.
Gavioli CFB, Nico MMS, Florezi GP, Lourenço SV. The histopathological spectrum of Melkersson-Rosenthal syndrome: Analysis of 47 cases. J Cutan Pathol. 2020; 47(11):1010-1017.
Wehl G, Rauchenzauner M. A systematic review of the literature of the three related disease entities cheilitis granulomatosa, oorofacial franulomatosis and Melkersson-Rosenthal Syndrome. Current Pediatric Reviews. 2018; 14:196-203.
Cappellini MD, Motta I. Anemia in Clinical Practice-Definition and Classification: Does Hemoglobin Change With Aging? Semin Hematol. 2015; 52(4):261-9.
Johnson-Wimbley TD, Graham DY. Diagnosis and management of iron deficiency anemia in the 21st century. Therapeutic Adv Gastroenterol 2011; 4:177-184.
Ganz T. Anemia of inflammation. N Engl J Med. 2019; 381:1148-57.
Briani C, Dalla Torre C, Citton V, Manara R, Pompanin S, Binotto G, Adami F. Cobalamin deficiency: clinical picture and radiological findings. Nutrients. 2013 15;5(11):4521-39.
Miller JW. Proton Pump Inhibitors, H2-Receptor Antagonists, Metformin, and Vitamin B-12 Deficiency: Clinical Implications. Adv Nutr. 2018; 1;9(4):511S-18S.
Kaur N, Nair V, Sharma S, Dudeja P, Puri P. A descriptive study of clinico-hematological profile of megaloblastic anemia in a tertiary care hospital. Med J Armed Forces India. 2018; 74(4):365-70.
Corresponding author: Karin Garming Legert, Department of Dental Medicine, Karolinska Institutet, PO BOX 4064, SE 141 04 Huddinge, Sweden. E-mail: karin.garming.legert@ki.se
Accepted for publication 11.10.2023. The article is peer reviewed.
Artikkelen er fagfellevurdert.
Artikkelen siteres som:
Legert KG, Pedersen AML, Gale G, Tollemar V, Ruokonen H, Kantola S, Wexell CL, Herlofson BB. Oral manifestations of systemic disorders – part 1. Nor Tannlegeforen Tid. 2024;134:104-13. doi:10.56373/2024-2-3