Bioinformatikk anvendt på lokalanestetikas virkningsmekanisme
Forfattere
førsteamanuensis, dr.odont. Odontologisk institutt – odontologisk farmakologi, UiB
seniorforsker/gruppeleder, PhD. Enhet for Beregningsorientert Biologi/Bergen Senter for Beregningsvitenskap, UiB
universitetsbibliotekar, cand.scient. Det matematisk naturvitenskapelige fakultetsbibliotek, UiB
Flere teknologiplattformer har i de senere årene blitt etablert for å styrke norsk forskning. En plattform er etablert i bioinformatikk, dvs. bruk av informasjonsteknologi til å forstå biologi. Verktøyet kan bl.a. brukes til å utvikle nye medikamenter, og det var en del av grunnlaget for introduksjon av legemiddelet sildenafil som Viagra®. Bioinformatikk kan også brukes til å studere basale farmakologiske prosesser. Et medisinsk, men også et viktig odontologisk diskusjonstema, er hvilke molekylære mekanismer som er ansvarlige for at lokalanestetika virker. I artikkelen presenteres en modell der matematisk modellering basert på kjemiske egenskaper gir muligheter for å studere dynamiske aspekter ved biologiske prosesser. Dette ble anvendt på artikain (Septokain®) og lidokain (Xylokain®) i vekselvirkning med cellemembranen. Funnene antyder at det kan være forskjeller i virkningsmekanismen til artikain og lidokain basert på kjemiske egenskaper. Resultatene vil bli fulgt opp i et samarbeid med bioinformatikk-plattformen ved Universitetet i Bergen ved bruk av molekylær dynamisk simulering.
FUGE bioinformatikk-plattform er en av elleve teknologiplattformer som ble etablert for å styrke norsk forskning innen funksjonell genomikk (FUGE). Bioinformatikkplattformen har som mål å drive forskning innen bioinformatikk på et internasjonalt nivå og å støtte norsk funksjonell genomforskning på bioinformatikk-siden. Plattformen koordineres av Universitetet i Bergen, Universitetet i Oslo og Norges teknisk vitenskapelige universitet i Trondheim og ledes av Enhet for Beregningsorientert Biologi (CBU) ved Universitetet i Bergen.
Tjenester fra CBU er basert på forskningsaktiviteter og består av to hovedelementer. Det ene omfatter utvikling av verktøy og databaser som gjøres allment tilgjengelig, som oftest gjennom websidene til plattformen (www.bioinfo.no). Det andre er direkte hjelp til brukere. Hjelpen kan bestå i alt, fra enkle råd om valg av metode eller programvare, til at plattformen gjennomfører bioinformatikkdelen av et stort prosjekt. Servicegruppen har etablert en «diagnosetjeneste». Dette betyr at etter at det er kommet et spørsmål per e-post, bruker gruppen en dag eller to på å analysere problemstillingen og gi brukeren en tilbakemelding om gjennomførlighet, estimat på tidsramme og eventuelt kostnader. Deretter vil brukeren inngå en avtale med servicegruppen, gjerne ved å møtes og diskutere nærmere konkrete handlinger. I tillegg organiserer plattformen flere kurs hvert år, rettet mot forskere og studenter (master eller PhD). Temaer kan være metoder i anvendt bioinformatikk (sekvensanalyse, proteinstrukturbestemmelse, analyse av mikromatrise-data, molekylær dynamikk), statistikk (grunnleggende statistikk) og forskjellige programmeringsspråk.
Bioinformatikk, beregningsorientert biologi og molekylær modellering
Det finnes flere ulike definisjoner av bioinformatikk. Et eksempel på en meget vid definisjon er «bioinformatikk er å bruke informasjonsteknologi til å forstå biologi». Det amerikanske nasjonale helseinstitutt (NIH) skiller mellom bioinformatikk og beregningsorientert biologi (computational biology) og definerer det sistnevnte som: «Utvikling og anvendelse av dataanalytiske og teoretiske metoder, matematisk modellering og metoder for beregningssimulering for å studere biologiske, atferdsmessige og sosiale systemer.»
Molekylær modellering er en metode for strukturelle studier av blant annet membraner og membrankomponenter basert på eksperimentell informasjon, i tillegg til bioinformatikk. Ved molekylær modellering forstår man kunnskapen å studere molekylær struktur og funksjon ved modellbygging og beregning (1).
Andre strukturelle og dynamiske metoder
Nukleær magnetisk resonans (NMR) og røntgenstrålediffraksjon er biofysikalske metoder som kan anvendes til å gi strukturell informasjon på molekylnivå. De brukes bl.a. til å beskrive den tredimensjonale (3-D) struktur av mindre molekyler, store biomolekyler, samt medikamenter og proteiner.
Molekylær modellering er komplementær til NMR og røntgendiffraksjon og kan brukes til å simulere biologiske molekyler i betingelser som reproduserer in vitro betingelser (temperatur, trykk, konsentrasjon). Dette betegnes som molekylær dynamisk simulering (MD). Metoden gir både strukturell og dynamisk informasjon.
Farmakologi og bioinformatikk
Utviklingen av medikamentet Viagra® ble resultatet av et forskningsprogram startet i 1985 for å utforme og syntetisere nye medikamenter for kardiovaskulære lidelser, bl.a. hypertensjon. Anvendelse av beregningsorientert biologi bekreftet i dette prosjektet at utgangsmedikamentet zaprinast hadde en tredimensjonal struktur som var forenlig med binding til enzymet phosphodiesterase. Resultatet av dette programmet ble imidlertid lanseringen av sildenafil som Viagra® i 1998.
Den basale virkningsmekanisme for de fleste medikamenter er via enzymer eller membranproteiner. Cellemembranproteiner omfatter ionekanaler og reseptorer som overfører signaler fra utsiden av membranen til innsiden. De er involvert i et utall av cellulære prosesser og utgjør et stort potensial som mål for nye medikamenter. For at medikamenter skal kunne virke i kroppen, må deres tredimensjonale struktur tilpasses kroppens egne stoffer. Dette er det basale grunnlaget for at det skal skje en vekselvirkning (interaksjon) i kroppen. Den tredimensjonale strukturen til mange reseptorer og enzymer er imidlertid ukjent. For å utvikle nye medikamenter er derfor detaljert strukturell informasjon viktig, både for medikamentet og kroppens bestanddeler.
Cellemembraner er svært sentrale i forståelsen av hvordan medikamenter virker i organismen. Funksjonelle membraner kan betraktes som todimensjonale væsker, og de er grunnlaget for farmakokinetiske og farmakodynamiske prosesser, dvs. enten medikamentet kun skal passere membranen eller at membranens lipider er målmolekylene. Modeller av cellemembraner kan bygges opp i datamaskiner ved hjelp av bioinformatikk. Et fosfolipidlag bestående av to fettsyrer og en vannløselig del (hodedel) som binder dem sammen, kan bygges opp på samme måte som i en biologisk membran, men basert på tilgjengelige strukturelle data (2). Både sammensetningen, type av fettsyre og hodedelen kan varieres og vil ha betydning for relevansen av modellen. For at modellen og stoffenes oppførsel i den skal være mest mulig realistisk, plasseres vannmolekyler på begge sider av membranen. Ved dynamisk simulering basert på parametisering kan man etterligne vekselvirkningen som skjer mellom legemiddelet og cellemembranen. Stoffene tilføres ladning, og krefter og vekselvirkningen observeres over tid. Dette stiller store krav til datamaskinens kapasitet, men også til valg av programvare. De fleste tilgjengelige programmer inneholder parametre for proteiner, i mindre grad for lipider.
Lokalanestetika og farmakologi
Strukturformlene til lokalanestetika indikerer amfifile egenskaper (dvs. at de inneholder både hydrofile og hydrofobe grupper) og de har således evnen til å interagere direkte med cellemembraner uten å gå veien om reseptorer.
Den basale molekylære virkningsmekanismen for lokalanestetika er under stadig diskusjon. Et antatt scenario er at stoffene diffunderer gjennom nervemembranen og gjenoppretter den kjemiske likevekten ved den indre del av nervemembranen. Deretter penetrerer lokalanestetika inn i membranen og binder til reseptorer i natriumkanalen. Om bindingsstedet for lokalanestesimiddelet utelukkende er en bestemt aminosyresekvens av proteinet eller deler av fosfolipider, eller begge deler, er imidlertid fremdeles diskutabelt (3). Ingen har hittil kunnet vise at en bestemt tredimensjonal aminosyresekvens i humane perifere aksonale natriumkanaler er forenlig med den tredimensjonale strukturen til eksempelvis lidokain. Det kan også tenkes at vekselvirkningen mellom lokalanestetika og cellemembranen kan ha betydning for den basale molekylære virkningsmekanismen til lokalanestetika. Slike vekselvirkninger vil også være av betydning ved transport av medikamenter gjennom membranen. Den nære relasjonen mellom proteiner og fosfolipider i cellemembranen gjør også at vekselvirkning med en type cellebestanddeler vil kunne påvirke den tredimensjonale strukturen til andre bestanddeler i membranen. Det er derfor gode grunner til å studere vekselvirkninger mellom membraner og medikamenter.
Molekylær dynamisk simulering av artikain og lidokain
Molekylær dynamisk simulering er blitt brukt til å studere forskjellige typer av lokalanestetika som vekselvirker med modellmembraner (4, 5). I disse undersøkelsene ble fosfolipider bestående av mettede fettsyrer, samt en hodedel av kolin, benyttet. Dobbeltbindinger i fettsyrene er kjent å kunne modifisere egenskapene i betydelig grad i cellemembranene. Et biologisk relevant fosfolipid bør derfor innholde flerumettede fettsyrer. I tillegg må molekylene tilføres ladning.
Et viktig poeng ved simuleringer er tidsaspektet. Dette har en klar sammenheng med hvilke fenomener som observeres. Hvis man vil studere diffusjonsprosesser (bevegelse av molekyler) vil man trenge lengre simuleringstid enn ved vekselvirkninger mellom molekyler (interaksjoner). Vekselvirkningene er grunnlaget for medikamentenes basale mekanisme. Eksempelvis vil intermolekylære vibrasjoner ha et tidsaspekt på 10–14 sekund. I de første publiserte arbeider med umettete fettsyrer ble simuleringer begrenset til kun å vare i 100 pikosekunder (10–10). Med dagens teknologi kan simuleringer kjøres i 100 nanosekunder(10 – 7). Prosesser som involverer lokalanestetika skjer svært raskt (4).
Lidokain er virkestoffet i Xylokain®. Artikain er virkestoffet i Septokain® og Septokain Forte®. Til tross for at den prinsipielle oppbygning av disse molekylene er like har de kjemiske egenskaper som er forskjellige. Dette kan ha betydning for den molekylære mekanismen.
For å konstruere en tredimensjonal modell av artikain ble substanser med tilnærmet lik kjemisk struktur anvendt (6 – 8) (Figur 1). Ved hjelp av tilgjengelige data (2) og programvare (Insight II) ble også en tredimensjonal struktur av et fosfolipidmolekyl modellert. Det ble da konstruert en monolagsmembran (dvs. ett lag fosfolipider) bestående av fosfolipider med vannmolekyler på begge sider. Programmet MOIL-View (9) ble brukt til å lage den molekylære grafikken. Etanolamin som hodedel i fosfolipidet representerer en modell av cellemembranens indre del, dvs. mot cytosol. Når artikain tilsettes membranen, kan man observere en dynamisk biologisk prosess i et modellsystem, dvs. at man kan observere at lokalanestesimolekylet beveger seg i nervecellens membran.
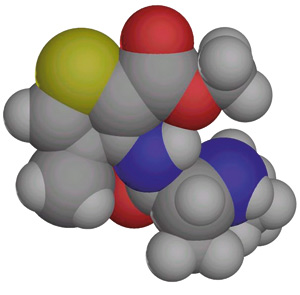
Figur 1. Modell av artikain. Den blå fargen er nitrogenatomer, gule er svovel, røde er oksygen, store grå er karbon og små grå er hydrogen.
I dette forsøket ble cellemembranene bygget opp av fosfolipider med fettsyrer av forskjellig lengde. De langkjedede fettsyrer viste bedre integreringsevne for lokalanestetika enn de korte. Ved sammenligning av artikain og lidokain viste artikain best integreringsevne i en monolagsmembran. Artikain var orientert slik at tiopenringen lå parallelt med fosfolipidets hodedel (Figur 2). Den andre delen av molekylet var orientert mellom fettsyrekjedene. I tillegg til disse strukturelle data er det også mulig, ved hjelp av denne metoden, å måle hvilke type av bindinger som skjer mellom atomer, samt i hvor lang tid bindingene varer.
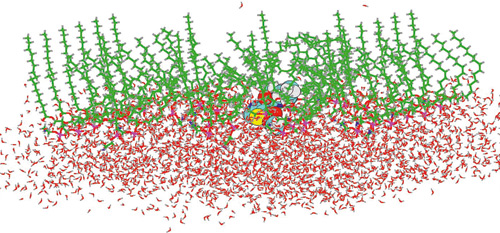
Figur 2. Øyeblikksbilde av molekylær dynamisk simulering applisert på artikain som vekselvirker med ettlags membran. Artikain ligger slik at tiopenringen i molekylet er parallell med fosfolipidets hodedel. De grønne «stengene» er fettsyrer.
Funnene i dette preliminære forsøket er, med sine begrensninger, en måte å visualisere farmakologiske prosesser på. De kan sannsynliggjøre forskjeller mellom den basale molekylære mekanismen for artikain og lidokain. Den informasjonen som foreligger med den omtalte metoden er ikke nok til å trekke en endelig konklusjon vedrørende lokalanestesimidlenes basale mekanisme. Funnene danner imidlertid et godt utgangspunkt for videre studier med NMR, men også for videreføring av den omtalte metoden. Et samarbeid med bioinformatikk-plattformen CBU ved Universitetet i Bergen er planlagt for å videreføre prosjektet.
Takk
Prosjektet er finansiert med midler fra Melzters Høyskolefond ved Universitetet i Bergen. Norsk Forskningsråd stilte midler til rådighet fra det norske programmet for forskning i FUnksjonell GEnomforskning (FUGE).
Hovedbudskap | |
|---|---|
• |
En bioinformatikk-plattform er etablert for å styrke norsk forskning innen funksjonell genomikk(FUGE) |
• |
Plattformen ledes av Enhet for Beregningsorientert Biologi (CBU) ved Universitetet i Bergen |
• |
Ved hjelp av bioinformatikk-verktøyet molekylær modellering kan man simulere biologiske/farmakologiske reaksjoner |
• |
Metoden er anvendt på artikain og lidokain for å studere deres molekylære virkningsmekanisme |
English summary
Lygre H, Reuter N, Rødland I.
Bioinformatics applied on the mode of action of local anesthetis
672 – 5.
In recent years platforms of technology have been established in Norwegian universities to strengthen the level of research. Among others, a platform of bioinformatics, i.e. the use informatics to understand biology, has been build. This kind of technology may be used in drug-design, as with the introduction of Viagra®. Basic pharmacological processes may also be studied by these methods. There is still a discussion about the molecular mechanisms in local anaesthetics. In this preliminary report a model by mathematical modelling based on chemical properties is presented. Applied on articaine (Septocaine®) and lidocaine (Xylocaine®) our data indicate that the molecular mechanism in these local anaesthetics may not be identical. Further research in cooperation with The Computational Biology Unit at the University of Bergen has been promoted to seek more conclusive findings.
Referanser
1. Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nat Struct Biol 2002; 9: 646 – 52.
2. Tieleman DP, Berendsen HJ. A molecular dynamics study of the pores formed by Escherichia coli OmpF porin in a fully hydrated palmitoyloleoylphosphatidylcholine bilayer. Biophys J 1998; 74: 2786 – 801.
3. Tsuchiya H, Mizogami M, Takakura K. Reversed-phase liquid chromatographic retention and membrane activity relationships of local anesthetics. J Chrom A 2005; 1073: 303 – 08.
4. Pasenkiewicz-Gierula M, Róg T, Grochowski J, Serda P, Czarnecki R, Librowski T, et al. Effects of carane derivative local anesthetic on phospholipid bilayer by molecular dynamics simulation. Biophys J 2003; 85: 1248 – 58.
5. Högberg CJ, Maliniak A, Lyubartsev AP. Dynamical and structural properties of charged lidocaine in a lipid bilayer. Biophys Chem 2007; 125: 416 – 24.
6. Hanson AW, Banner BW. 2-Diethylamino-2",6"-acetoxylidide (lidocaine). Acta Cryst 1974; B30: 2486 – 88.
7. Weber L. The application of multi-component reactions in drug discovery. Curr Med Chem 2002; 9: 1241-53.
8. Storsberg J, Schollmeyer D, Ritter H. Route towards new heteroatomic benzo[1,4] dioxine derivates. Chem Lett 2003; 32: 140.
9. Simmerling C, Elber R and Zhang J. MOIL-View – A Program for visualization of structure and dynamics of biomolecules and STO- A Program for computing stochastic paths. In: Pullman A et al., editors. Modelling of biomolecular structure and mechanisms. Netherlands: Kluwer; 1995.p.241 – 65.
Adresse: Henning Lygre, Seksjon for farmakologi, Armauer Hansens Hus, 5021 Bergen. E-post: henning.lygre@odont.uib.no
Artikkelen har gjennomgått ekstern faglig vurdering.