Orale manifestasjoner ved autoimmunt polyendokrint syndrom type 1
Forfatterbidrag
Hanne Borge: Har skrevet artikkelen og bidratt med faglige råd og design samt utarbeiding av retningslinjer av sykdomskjennetegn, behandling og oppfølging av oral helse hos pasienter med APS-1.
Cecilie Gjerde, Kathrine Skarstein, Hilde Nordgarden: Har bidratt med kliniske og patologiske undersøkelser av pasienter med APS-1, faglig kompetanse i behandling og vurdering av kasusene samt veiledning av masterstudenter.
Athanasia Bletsa, Siri Flagestad Kvalheim: Har bidratt med kliniske undersøkelser av pasienter med APS, faglig kompetanse i vurdering og behandling av pasientene.
Artikkelen er delvis basert på to prosjektoppgaver ved Integrert master i odontologi, Institutt for klinisk odontologi, Universitetet i Bergen (Ellen Guddal, Hanne Borge, Andrea Runningen og Abhira Sivakumar).
Eystein S. Husebye, Anette S. B. Wolff, Mihaela Cuida Marthinussen: Har bidratt med lik tverrfaglig kompetanse innen APS-1 med oppfølging av pasienter over lang tid og spesialkompetanse på forskningsområdet og i behandling og diagnostisering av pasientene.
Autoimmunt polyendokrint syndrom type 1 (APS-1) er en sjelden autoimmun organspesifikk sykdom som skyldes recessiv arv. Sykdomsmanifestasjonene er primær binyrebarksvikt (Addisons sykdom), svikt i biskjoldbruskkjertelen (hypoparatyreoidisme) og kroniske soppinfeksjoner. Sykdomsbildet varierer fra individ til individ, hvor både endokrine og ikke-endokrine vev er affisert med sykdomsdebut i barneårene, men med diagnosetidspunkt oftest senere i livet. Pasientene har flere orale manifestasjoner som emaljedefekter hvor alvorlighetsgraden varierer, fra mindre hypomineraliseringer til uttalte hypoplasier. Vi antar at emaljedefekter ved APS-1 er underdiagnostisert. Mange pasienter med APS-1 utvikler tidlig i livet kroniske soppinfeksjoner forårsaket av Candida albicans i munnslimhinnen og øsofagus, noe som øker risikoen for dysplasi og patologiske forandringer. Sårdannelser og atrofi av munnslimhinnen er vanlige funn.
Grunnet de orale manifestasjonene hos pasienter med APS-1 er det viktig at tannhelsepersonell har kjennskap til sykdomskarakteristika ved syndromet. Dette vil kunne gi bedre diagnostikk og behandling for pasientene i et tverrfaglig behandlingsteam som inkluderer endokrinolog, fastlege og tannlege. Ved mistanke om APS-1 basert på munnhulefunn eller ved en etablert diagnose kan pasienten henvises til et regionalt kompetansesenter, Nasjonalt senter for sjeldne diagnoser, enhet munnhelse (tidligere TAKO-senteret) eller en spesialistklinikk ved universitetene.
Hovedbudskap
Utredning av emaljeutviklingsforstyrrelser er viktig da det kan foreligge en alvorlig bakenforliggende sykdom, som APS-1, cøliaki og amelogenesis imperfecta.
Emaljeutviklingsforstyrrelser er en av de tidligste manifestasjonene ved APS-1 og skyldes autoimmunitet mot proteiner som inngår i emaljedanningen.
Tidlig undersøkelse og regelmessig oppfølging er viktig for å hindre ytterligere emaljeskader hos pasienter med APS-1.
Soppinfeksjoner hos pasienter med APS-1 må behandles for å hindre utvikling av slimhinneforandringer.
Autoimmunt polyendokrint syndrom type 1 (APS-1), også kjent som «autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED)», er en organspesifikk autoimmun sykdom kjennetegnet ved immunmediert infiltrasjon og destruksjon av vev, som gradvis fører til funksjonstap i de berørte organene. Sykdommen skyldes mutasjoner i genet som koder for proteinet «autoimmun regulator (AIRE)». Hovedmanifestasjonene er primær binyrebarksvikt (Addisons sykdom), svikt i biskjoldbruskkjertelen (hypoparatyreoidisme) og kroniske soppinfeksjoner, hvor minst to av disse manifestasjonene må være til stede for å stille diagnosen [1]. Sykdomsbildet varierer mye fra pasient til pasient med varierende kjertel- og organmanifestasjoner der både endokrine og ikke-endokrine vev er affisert (figur 1) [2]. Eksempler på dette er alopecia, vitiligo og autoimmun gastritt/pernisiøs anemi [2]. Det varierer også når de ulike komponentene av APS-1-syndromet opptrer i alder.
Figur 1
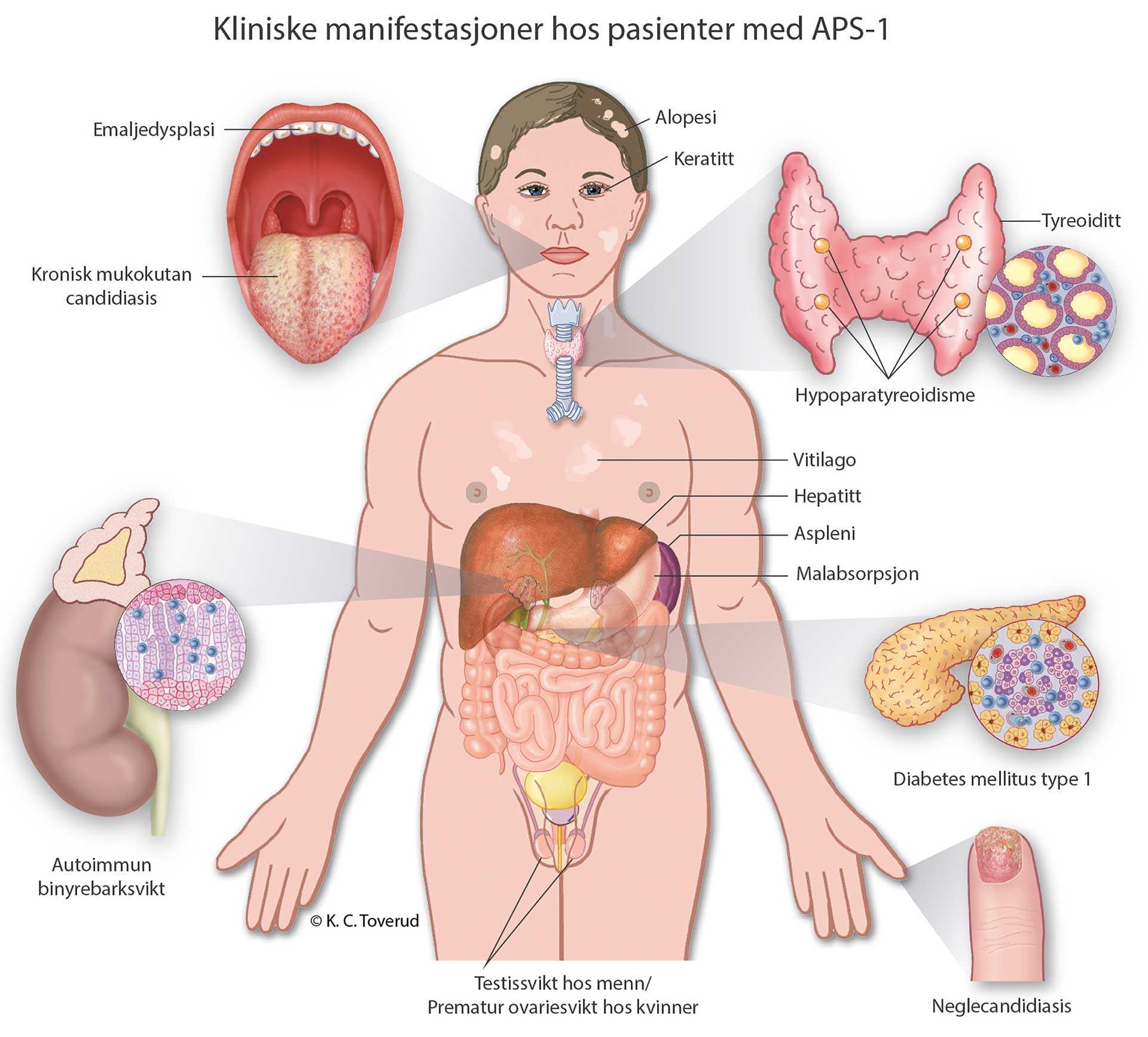
Figur 1. Kliniske manifestasjoner ved APS-1. Figur brukt med tillatelse fra Kari C. Toverud, MS CMI (certified medical illustrator).
Pasienter med APS-1 har ofte flere orale manifestasjoner der emaljehypoplasi og kroniske soppinfeksjoner er de hyppigste funnene. Utviklingsforstyrrelser i emaljen kan være det første tegnet på tilstanden, og det er derfor viktig at tannhelsepersonell har kjennskap til sykdomskarakteristika ved APS-1. Det vil kunne gi bedre diagnostikk og behandling for pasientene i et tverrfaglig behandlingsteam som inkluderer endokrinolog, fastlege og tannlege.
APS-1 er en sjelden sykdom med antatt prevalens på 1:100000 i Norge [3]. I enkelte befolkningsgrupper er frekvensen høyere, som i Finland (4:100000) [1], Sardinia (7:100000) [4] og blant persiske jøder (10:100000) [5]. Det er ingen forskjell i prevalens mellom kvinner og menn [3]. Noen land har etablert biobanker og registre for pasienter med APS-1, bl.a. i Finland [1], Russland [6], Sardinia [7], Italia [8][9] og Norge [3][10]. Dette informasjonsgrunnlaget er sentralt i forskningen på APS-1 og kan gi grobunn for ytterligere kunnskap om årsaksmekanismen og tilstedeværende sykdomsmanifestasjoner. I Norge kjenner man til om lag 50 personer med APS-1 [10].
Etiologi for APS-1
Arvegangen for klassisk APS-1 er autosomal recessiv (nedarvet fra både mor og far), men dominant arvegang (nedarvet fra en av foreldrene) er beskrevet, noe som ofte gir et mildere sykdomsforløp [11][12][13][14][15]. Årsaksgenet for APS-1 koder for proteinet autoimmun regulator (AIRE). AIRE-genet befinner seg på kromosom 21 (21q22.3), og flere enn 100 ulike mutasjoner er identifisert [16]. Proteinet AIRE hjelper immunforsvaret med å lære hva som er kroppens egne proteiner. I thymus viser AIRE frem slike proteiner til T-cellene mens de modnes. Da blir T-celler som kan angripe kroppen, enten fjernet eller gjort om til regulerende T-celler [17]. Hvis AIRE ikke virker, slipper farlige T-celler ut i blodet og kan senere gi autoimmun sykdom. Dette påvirker også B-cellene, som kan lage antistoffer mot kroppens egne molekyler [18].
Hos pasienter med APS-1 finner man ofte antistoffer mot affiserte vev i serum; eksempelvis mot 21-hydroksylase (21OH), 17-hydroksylase (17OH) og sidekjedespaltningsenzym («side-chain cleavage enzyme (SCC)») ved binyrebarksvikt og NACHT leucine-rich-repeat-protein 5 (NALP-5) ved hypoparatyreoidisme. Pasienter med APS-1 har også autoantistoffer mot interferon type 1 og flere interleukiner (IL) [10].
Orale manifestasjoner ved APS-1
Emaljeutviklingsforstyrrelser
Emaljehypoplasi er den vanligste orale manifestasjonen ved APS-1, som kan sees på flere tenner med ulik alvorlighetsgrad [19]. I litteraturen blir emaljedefekter fremhevet som et av de mest fremtredende ektodermale manifestasjonene av APS-1 [16]. Mønsteret og alvorlighetsgraden til emaljedefekter varierer fra pasient til pasient, fra mindre hypomineraliseringer til uttalte hypoplasier. Vi antar at dette er underdiagnostisert [20][21]. I en longitudinell studie utført av Bruserud et al. ble det rapportert emaljehypoplasier hos 72 % av norske pasienter med APS-1 [10]. Lignende frekvens ble også observert av Ferre et al., som undersøkte en amerikansk kohort. Der fant de at 85,7 % hadde emaljehypoplasi og at dette også var en av de tidlige manifestasjonene på syndromet [22].
Flere ulike hypoteser er blitt fremsatt for hvorfor tannemaljen blir ødelagt hos pasienter med APS-1. Emaljehypoplasi ved APS-1 kan være forårsaket av tidlig debut av hypoparatyreoidisme, en utviklingsdefekt der AIRE er direkte involvert i danningen av emaljen, eller på grunn av en autoimmun reaksjon mot proteiner som er essensielle for emaljedanning. Det er viktig å kartlegge når emaljedefektene har oppstått ved å se på kronologien i tanndanningen, for å se om defektene har sammenheng med tidspunkt for tidlig sykdomsdebut eller genetiske betinget feil i utviklingen [23]. Emaljedefektene man finner hos pasienter med APS-1, korresponderer med gitte perioder under emaljedanningen, noe som taler imot en primær utviklingsdefekt forårsaket av mangel på AIRE-proteinet [24]. En autoimmun prosess mot ameloblast-antigener involvert i tanndanningen, som ACPT, FAM20A, AMELX, ENAM og LAMB3, er derimot kompatibelt med studier som viser hvor på tennene, og på hvilke tenner, og med hvilket mønster, APS-1-pasienter har sine emaljedefekter [21][24]. Gruper et al rapporterte nylig at pasienter med APS-1 med alvorlige emaljedefekter har høyere nivå av autoantistoffer mot flere ulike emalje-autoantigener, sammenlignet med pasienter med mildere emaljedefekter og friske [24]. Dette støtter den autoimmune hypotesen for utviklingsforstyrrelser av emaljen hos pasienter med APS-1.
Kronisk soppinfeksjon
Over 80 % av pasientene med APS-1 utvikler kroniske soppinfeksjoner forårsaket av Candida albicans i slimhinner tidlig i livet [2][25]. Av disse har alle orale soppinfeksjoner, mer enn 50 % har spiserørs- og vulvovaginale infeksjoner, og det er rapportert om varierende grad av neglesopp, som også er blitt forklart som negledefekter [20][22]. Det er rapportert om uttalte soppinfeksjoner i munn/svelg uten store utslag på dyrkningsprøver, og det er også forfatternes erfaring at kliniske indikasjoner på sopp i slimhinner ikke alltid stemmer med laboratorieprøvene og i enkelte tilfeller kan skyldes andre infeksjoner med bakterier og/eller virus [26]. Utvikling av plateepitelkarsinom i munn/tunge er en risikofaktor ved kroniske soppinfeksjoner [1][27], og det er derfor viktig å følge opp APS-1-pasienter med årlige slimhinneundersøkelser.
Vi kan dele soppinfeksjonene inn i to undergrupper, atrofiske og hyperplastiske candidainfeksjoner [18]. Orale soppinfeksjoner varierer fra en mildere form lokalisert i munnvinkelen, til en hyperplastisk infeksjon som påvirker hele munnhulen [28]. Pasienter med APS-1 kan ha begge typer og hele spekteret fra milde infeksjoner til affeksjon av store deler av munnhulen. Hyperplastisk soppinfeksjon karakteriseres med et grålig og hvitlig belegg på slimhinnen og hyperkeratose primært lokalisert på tungen og mucosa [28]. Den atrofiske formen kjennetegnes ved erytematøse slimhinneforandringer [18].
Slimhinneforandringer
Pasienter med APS-1 er mer utsatt for utvikling av dysplasi og forandringer i munnslimhinnen som på sikt kan utvikles til plateepitelkarsinom, både i munnslimhinnen og slimhinnen i øsofagus [1][10][27]. RNA-sekvensering har ikke påvist korrelasjoner mellom AIRE-genuttrykket og plateepitelkarsinomutvikling [29][30]. Man tror likevel at AIRE kan ha en rolle i utviklingen fra kronisk oral candidainfeksjon til dysplasi og plateepitelkarsinom hos APS-1, men at mekanismene kan være sekundære. Mye er fortsatt ukjent ved de molekylære mekanismene bak oral karsinomutvikling hos noen pasienter med APS-1.
Munntørrhet
Mer enn 40 % av pasientene med APS-1 har enten øyemanifestasjoner og/eller tørr munn (xerostomi) som ligner manifestasjonene ved Sjögrens sykdom [31]. Laboratoriemålinger som støtter hyposalivasjon og redusert tåredanning foreligger, men pasientene med APS-1 har ikke tilstedeværelse av de samme autoantistoffene som ved Sjögrens syndrom [20][31]. Munntørrhet kan føre til såre slimhinner, svie i munnen, forsinket sårtilheling, rask utvikling av karies og erosjoner samt soppinfeksjoner i munn og svelg. Pasientene kan oppleve vansker med spising og svelging og endret smaksoppfatning. Andre problemer kan være tørre lepper, cheilitis og talevansker [31].
Andre kormorbiditeter hos pasienter med APS-1
Pasienter med APS-1 kan også ha andre tilstander som tannleger bør være oppmerksomme på [10]. Det kan være sår i munnhulen, neglesopp, vitiligo (pigmentforandringer), alopecia (håravfall), hyperpigmentering hud/slimhinner (vanlig ved binyrebarksvikt) og generell fatigue koblet til annen helseproblematikk [10].
APS-1 – Kasuistikker
Vi beskriver her tre kliniske kasus for å skissere oral helseproblematikk hos pasienter med APS-1.
Pasient 1
Dette er en pasient som ble diagnostisert med APS-1 ved ettårsalder. Hen har alle tre hovedkomponentene av APS-1: hypoparatyreoidisme, binyrebarksvikt og kroniske candidainfeksjoner, og behandles med substitusjon av hormoner. Pasienten ble også diagnostisert med uttalt Candida oesofagitt og tidvis Candida stomatitt. Andre komponenter pasienten har, er malabsorpsjon og keratitt. Pasienten ble henvist fra Haukeland universitetssjukehus til munnundersøkelse med tanke på emaljehypoplasi. Pasienten har fått bekreftet diagnosen med påvisning av homozygositet for AIRE-mutasjon. Autoantistoffer som ble funnet i serum fra pasienten, inkluderer antistoffer mot IFN-omega, 17OH og IL-22.
Faste medikamenter: Hydrokortison, Rocaltrol, Florinef, Nycoplus magnesiumtabletter, Nexium 40 mg.
Kjente allergier: Ingen
Intraoral undersøkelse: Normale slimhinner uten tegn til candidainfeksjon. Flere tenner hadde komposittrestaureringer. Det var også tydelig tannslitasje i form av erosjon og abrasjon på flere tenner. Hypoplastiske emaljedefekter kunne sees på flere tenner i alle kvadranter. Tann 23 har slitasje til dentin. Det registreres hypoplasi, men for noen av disse defektene kan posteruptive brekkasjer ikke utelukkes. Hypoplasi ble registrert bukkalt/okklusalt på tann 14, 16, 22, 24, 25, 26, 33, 32, 31, 41, 42, 43 (figur 2). Hypoplasi lingualt i form av furer ble observert på tann 33 og 43. Multiple kariesangrep ble også diagnostisert, men kun symptomer fra tann 16 med behov for endodontisk behandling.
Figur 2
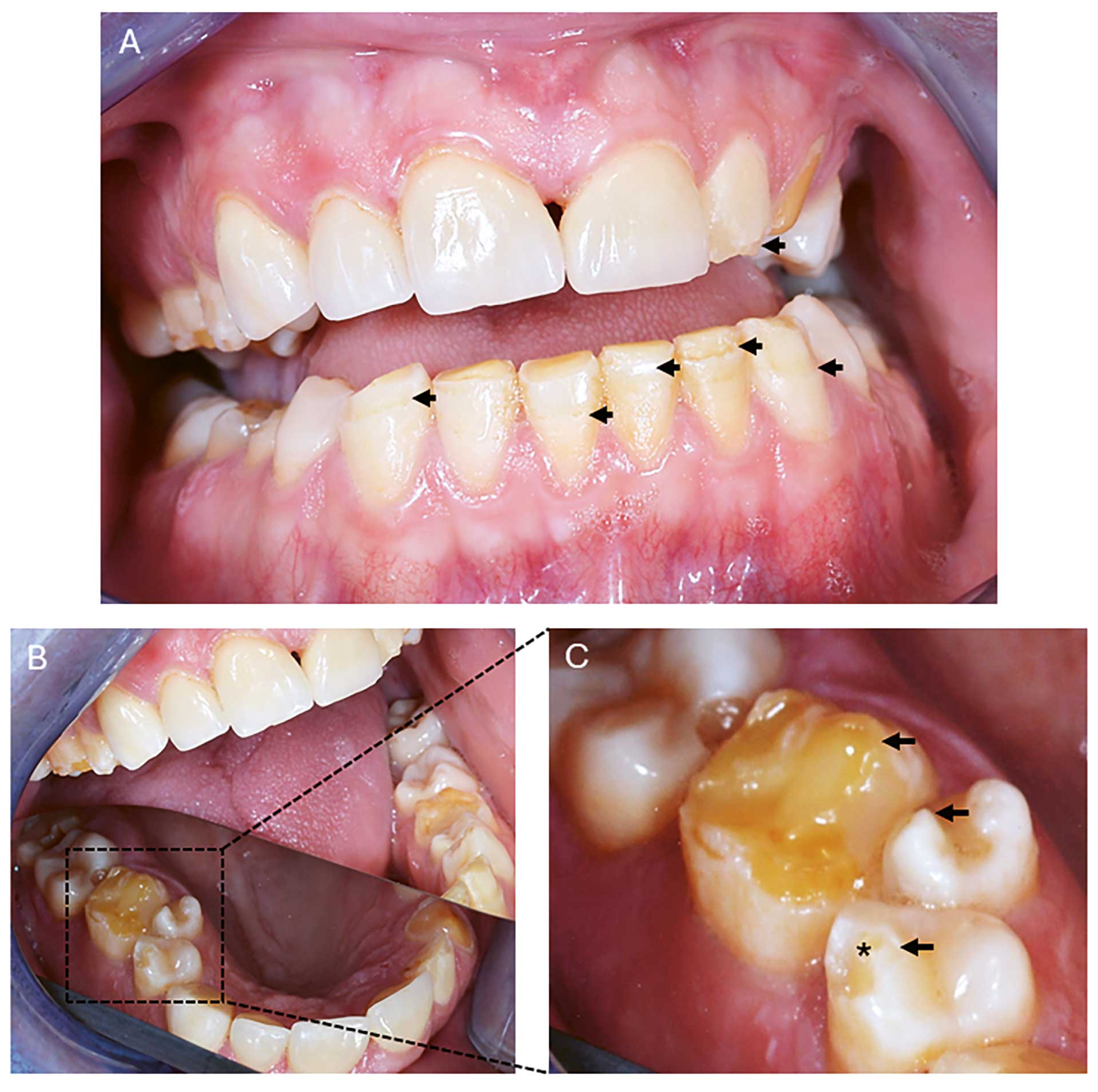
Figur 2. Klinisk foto av orale manifestasjoner hos pasient 1. (A) Emaljehypoplasi kan sees på 22, 31, 32, 33, 41 og 43 (svarte piler) (B) Oversiktsbilde av 1. kvadrant ved hjelp av speil (stiplet område). (C) Større forstørrelse av tann 16, 15 og 14 i B med emaljehypoplasi (svarte piler) og 14 med fraktur av emalje på kuspetopp (stjerne).
Pasient 2
Dette er en pasient med APS-1 som først ble diagnostisert med primær binyrebarksvikt ved tiårsalder, men med symptomdebut for kronisk candida i munnhulen allerede før fylte ett år. AIRE-mutasjoner ble påvist, og pasienten har også andre APS-1-manifestasjoner, som kronisk hepatitt. Autoantistoffer mot 21OH og IFN-omega er funnet i oppfølgingstiden. Pasienten har brukt natriumfluoridtabletter og hatt brønnvann i oppveksten, noe som er en risiko for dental fluorose da fluoridnivå i brønnvann trolig er høyere enn annet drikkevann. Pasienten har også gått regelmessig til undersøkelse hos offentlig tannhelsetjeneste. Ved undersøkelse informerer pasienten om en metallisk smak i munnen, og det kommer også frem at pasienten har vært plaget med gjentatte orale candida-infeksjoner over flere år (nesten kontinuerlig) samt at hen også er plaget med angulær cheilitt. Pasienten har hatt candidainfeksjon på fingerneglene.
Faste medikamenter: Kortisol, Floringe, Rocatrol (Vitamin D3), Diflucan, Creon, magnesium, kalium, kalsiumtilskudd og magnesiumtilskudd.
Intraoral undersøkelse: Lettblødende gingiva og tunge. Tungen er også lobulert. Det er registrert mild dental fluorose, hypomineralisering og emaljehypoplasi av tenner (figur 3). Flere tenner har komposittrestaureringer,og tann 25 har krone. Hypoplasi er registrert på tann 13-23, 16, 17, 26, 27, 28, 33, 36, 43, 46.
Emaljehypoplasi i underkjeven på 33, 43 bukkalt samt linguale furer på 33, 31, 42 og 43. Røntgenundersøkelsen viste mindre opakemalje enn normalvariasjoner i overkjeve front 13-23 samt underkjeve front. Saliva sekresjonstest viste 0,16ml/min ustimulert saliva og 1ml/min stimulert saliva med lav bufferkapasitet. Til sammenligning er verdier under 0,1 ml/min betraktet som patologisk, og mellom 0,1–0,25 som lav sekresjon [32].
Figur 3
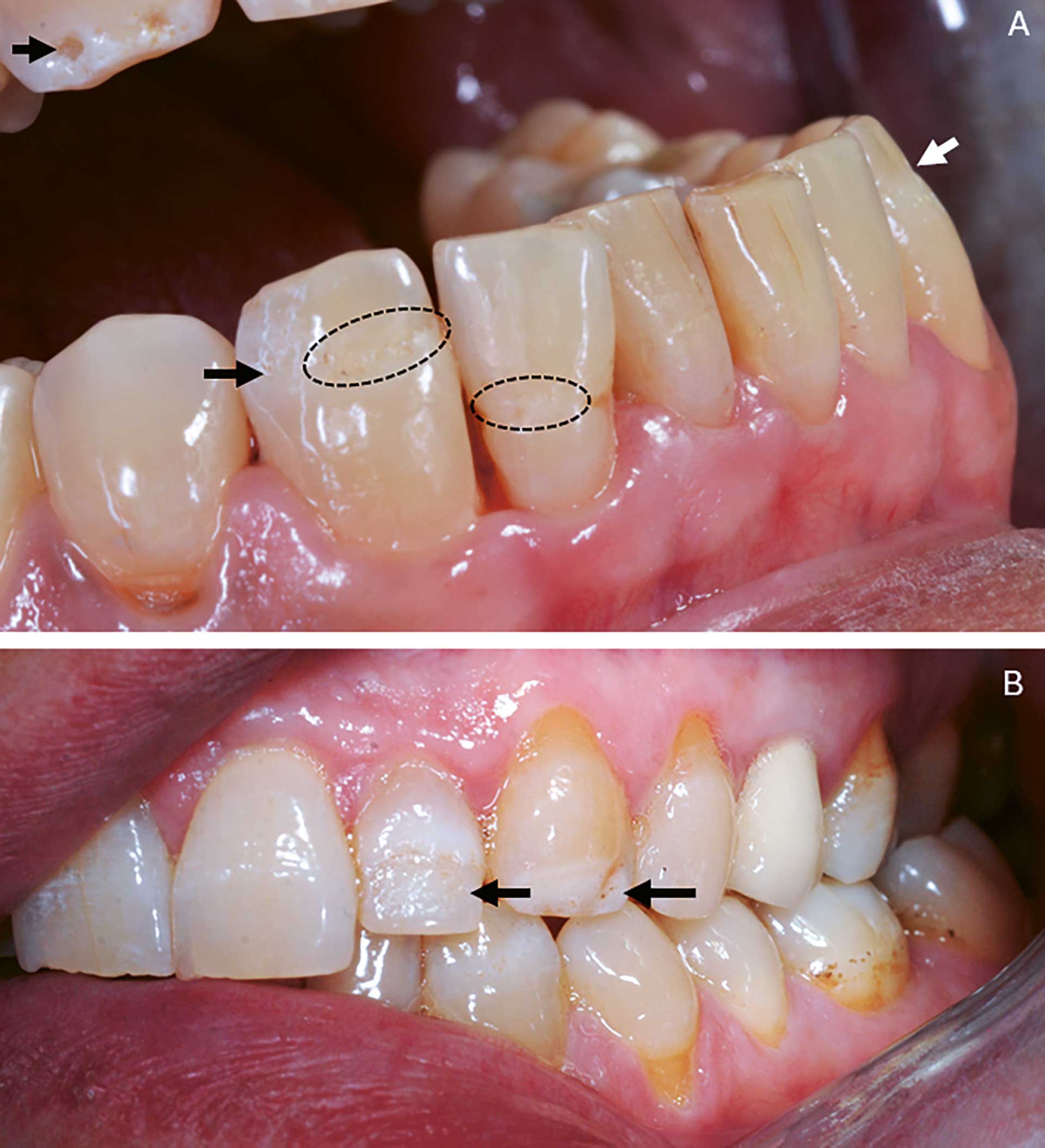
Figur 3. Kliniske foto av pasient 2 diagnostisert med APS-1. A: De intraorale funnene viser emaljehypoplasi bukkalt på tann 43 og 42 med innbuktninger på overflaten som etter en strikk (stiplet sirkel) og groper incisalt på tann 13 (pil). Innbuktningen av emaljeoverflaten sees i profil på tann 33 (hvit pil). B: Tann 22 med hypoplastisk emalje (pil) hvor den ujevne emaljen sees som spraglete og ru på buccalflaten. Tann 23 med innbuktning på emaljeoverflaten (pil).
Pasient 3
Pasienten ble diagnostisert med Addisons sykdom i en alder av 23 år. Samme år ble pasienten diagnostisert med soppinfeksjon på negl som senere ble kategorisert som negledystrofi (figur 4). Pasienten hadde en kjent arvelig predisposisjon ettersom ett av søsknene allerede var diagnostisert med APS-1, og pasienten fikk også påvist kronisk oral candidainfeksjon. AIRE-mutasjoner ble påvist i begge alleler. Hen hadde flere ulike autoantistoffer som er vanlige ved ulike komponenter ved APS-1-syndromet (21OH- og 17OH-autoantistoffer som korreleres med binyrebarksvikt, og NALP5-rettede antistoffer som er vanlig ved hypoparatyreoidisme). I tillegg fant man autoantistoffer mot IFN-omega.
Faste medikamenter: Hydrokortison, Fludrokortison, Calcichew/Vitamin D (1 tablett daglig).
Intraoral undersøkelse: Pasienten har kun påvisbare defekter på 11, 13, 31. Tann 11 var restaurert med kompositt på grunn av emaljedefekt incisalt og bukkalt. Ingen periodontal sykdom eller aktive kariesangrep ble registrert. Ustimulert og stimulert salivamåling ble utført med normale funn. Slimhinneprøve for Candida albicans viste ingen vekst.
Figur 4
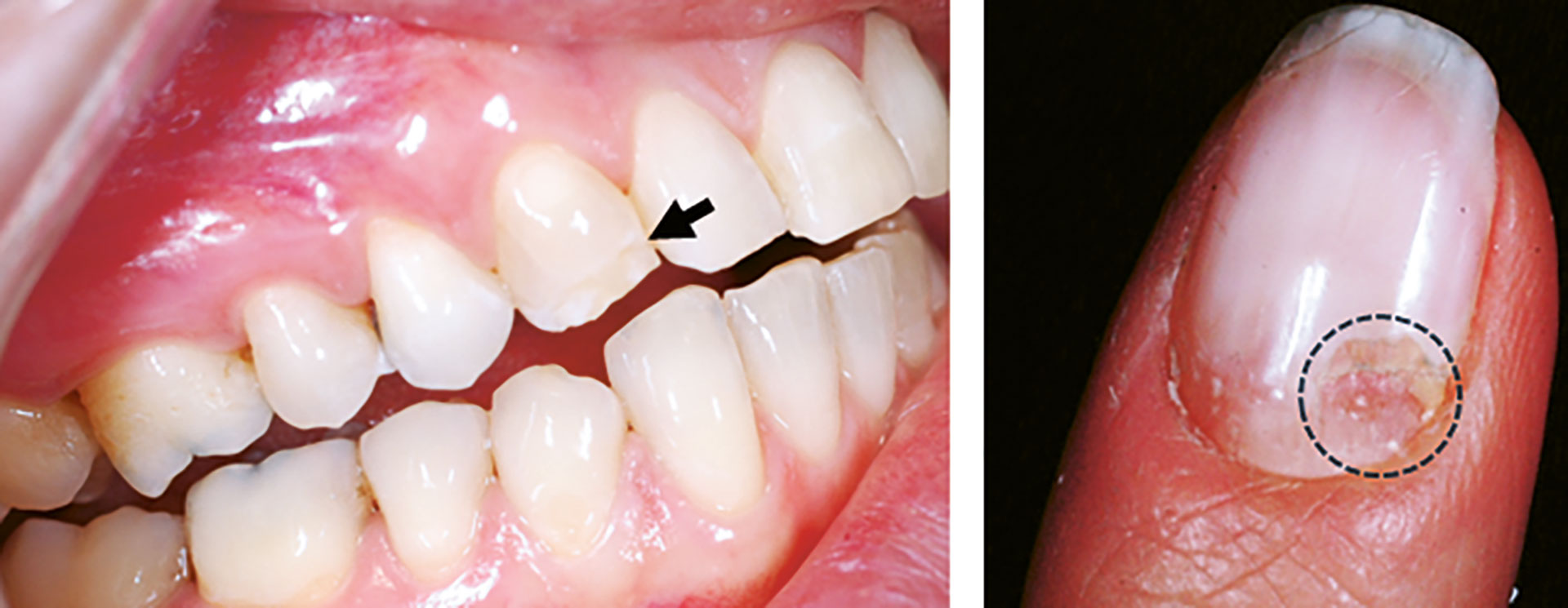
Figur 4. Intraoralt foto av hypomineralisert emalje (pil) og klinisk foto av negl med negldystrofi (stiplet område). Denne pasienten har få orale manifestasjoner av APS-1, men kan her sees på tann 13 med hypomineralisert emalje (pil). Pasienten har i tillegg forandringer på negl med groper og avskalling som er forenelig med APS-1. Forandringen ble først diagnostisert som soppinfeksjon før det ble rediagnostisert som negldystrofi (stiplet sirkel).
Anbefalt behandling og oppfølging
Generelt terapiforslag til pasienter med APS-1: Vi anbefaler pasientene å bruke natriumfluorid-tannkrem uten såpestoffet natriumlaurylsulfat (SLS), hjemmeprofylakse med 0,2 % natriumfluorid skyllevæske samt hyppig kontroll hos tannlege/oralkirurg. Det er viktig at pasienten får tillit til behandlende tannlege slik at karies, soppinfeksjoner og annen tannbehandling blir gjort så tidlig som mulig.
Appendiks 1 og 2 oppsummerer henholdsvis anbefalinger om syndromet for tannleger (appendiks 1) samt angir et skjema som kan brukes for undersøkelse av pasienter med APS-1/under utredning for APS-1 (appendiks 2).
Organisering: For å kunne ivareta denne pasientgruppen på en tilfredsstillende måte kreves det et tverrfaglig samarbeid mellom sykehus, tannhelsetjeneste og fastlege [33]. De medisinske tilstandene blir ivaretatt av endokrinologiske poliklinikker ved de store sykehusene i Norge, men ikke-endokrine manifestasjoner krever vanligvis henvisning til andre spesialister, som hudleger, gastroenterologer, infeksjonsleger, øre-nese-hals-spesialister og lunge- og øyeeksperter. Pasienter der man mistenker APS-1 basert på munnhulefunn eller som har en etablert diagnose, kan henvises til Nasjonalt senter for sjeldne diagnoser, enhet munnhelse (tidligere TAKO) ved Lovisenberg diakonale sykehus, til et av de regionale odontologiske kompetansesentrene eller til spesialisttannklinikker ved universitetene. Det er spesielt viktig at spesialist i oralkirurgi og oralmedisin er en del av teamet rundt pasientgruppen med tanke på oppfølging av slimhinneforandringer. Nasjonalt senter for sjeldne diagnoser, enhet munnhelse, har særlig kompetanse på orale manifestasjoner av sjeldne diagnoser og kan bidra i utredning og eventuell plan for behandling og oppfølging i samarbeid med de som har ansvaret for pasientene, enten lokalt eller ved et regionalt kompetansesenter. APS-1 står på A-listen over sjeldne medisinske tilstander, og pasienten har derfor krav på stønad til nødvendig tannbehandling [34]. Ved de nasjonale tannhelsekompetansesentrene har man erfaring med behandling og oppfølging av pasienter med sjeldne medisinske tilstander, og alle tannleger kan kontakte regionale kompetansesentre for råd og veiledning, og eventuelt behandling [35]. Pasienter med APS-1 har også økt risiko for kariesutvikling ettersom noen antimykotiske legemidler inneholder sukker i ulike former, for eksempel Nystatin mikstur. Dette kan predisponere pasienten for kariesutvikling på grunn av høyt sukkerinnhold i det antimykotiske middelet, kombinert med lav saliva sekresjon [36]. Det er viktig at allmenntannleger er observante på også disse tilstandene [35]. Det er videre viktig at pasientene blir anbefalt en tannkrem uten SLS, ettersom pasientene er munntørre og ofte har flere sår i slimhinnene som gjør at de lettere får ytterligere sårdanninger [37]. Tannlegene bør også ta kontakt med register for organspesifikke autoimmune sykdommer (ROAS) ved Haukeland universitetssjukehus, som er spesialister på oppfølging av APS-1, ved mistanke om tilstanden hos pasienter. Leder for ROAS er overlege/professor Eystein S. Husebye. AIRE-mutasjonsanalyse blir foretatt ved indikasjon ved Avdeling for medisinsk genetikk, Haukeland universitetssjukehus.
Diskusjon
I denne oversiktsartikkelen har vi illustrert hvor viktig tannlegens rolle er i tidig diagnostisering og oppfølging av APS-1. Dette er en sjelden diagnose som har varierende sykdomsdebut og ulikt symptombilde hos de enkelte pasientene. På grunn av de varierende sykdomsmanifestasjonene foreligger det trolig en underdiagnostisering [33]. Dette betyr at det er viktig at man som tannhelsepersonell kjenner til og er observant på de sentrale orale kjennetegnene av tilstanden. Da kan man bidra til at APS-1 kan diagnostiseres og at dokumentasjon og etablering av oppfølgingsregime til pasientene etableres på et tidlig stadium. Tannhelsepersonell er sentrale i alle faser av sykdommen, og det er essensielt at pasientene føler seg ivaretatt og oppnår tillit til behandlende tannlege.
Pasientene gjennomgår mange undersøkelser årlig, og flere av pasientene opplever tretthet knyttet til sykehus og helsepersonell. APS-1 har påvirkning på livskvaliteten til de som bærer syndromet og har i stor grad sammenheng med alvorlighetsgraden av sykdomsbildet [33][38]. Flere har binyrebarksvikt, hypoparatyreoidisme, kroniske soppinfeksjoner, diabetes mellitus og mangeltilstander som sammen gir en stor belastning for den enkelte [33]. Dette kombinert med redusert livskvalitet gjør også at neglisjering av egen oral helse kan forekomme [39]. Et viktig punkt er at annen medisinsk behandling kan bli prioritert over tannbehandling, noe som igjen kan føre til redusert oral helse. Pasienter med APS-1 bør ha hyppige innkallingsintervaller til tannhelseundersøkelse, og ved syndromrelaterte soppinfeksjoner bør pasienten følges opp etter et individuelt innkallingsregime. Det bør også være lav terskel for å henvise pasientene videre dersom det er mistanke om slimhinneforandringer [35].
Den diagnostiske utfordringen ved APS-1 ligger i stor grad i at de dentale funnene har betydelige likhetstrekk med andre emaljedefekter, enten disse er genetisk betinget, relatert til miljøfaktorer eller sekundære til sykdom. For å øke diagnostisk presisjon er det derfor nødvendig å supplere kliniske funn med radiologiske undersøkelser [23]. APS-1 har også kliniske likhetstrekk med andre sykdommer som gir hyposalivasjon, xerostomi og orale soppinfeksjoner, men med ulik etiologi [23][40]. Både emaljeutviklingsforstyrrelser som sees ved molar incisiv hypomineralisjon (MIH) og ved amelogenesis imperfecta kan være påfallende like APS-1-manifestasjoner [23][41]. Typisk for APS-1 er imidlertid at emaljeforandringene ofte er generaliserte, affiserer flere permanente tenner og forekommer sammen med mukokutan candidiasis (CMC) og endokrine dysfunksjoner, noe som skiller tilstanden fra isolerte dentale lidelser. Dental fluorose deler også flere fenotypiske trekk med både APS-1 og MIH, men har en distinkt etiologisk bakgrunn [40]. Munntørrhet, i form av hyposalivasjon og xerostomi, er ikke et spesifikt kjennetegn ved APS-1, men kan opptre sammen med andre orale patologiske funn [20][31]. Oftedal et al fant at 62 % av pasientene med APS-1 i en norsk studie (n=41) hadde spyttkjerteldysfunksjon med lav spyttproduksjon som konsekvens. Det ble også observert nedsatt funksjon i tårekjertlene med redusert tåreproduksjon i samme studie [31].
APS-1 har flere fellestrekk med emaljeforandringer sett hos pasienter med cøliaki. Man har undersøkt disse to sykdommene for autoimmun amelogenesis imperfecta, ettersom begge sykdommene har påvist autoantistoffer mot ameloblastspesifikke proteiner som amelogenin, som igjen påvirker amelogenesen [24]. Det er vist at emaljedefektene hos paseinter med APS-1 kan korreleres med emaljespesifikke autoantistoffer i barneårene og tidsforløpet emaljen er eksponert for disse autoantistoffene under mineraliseringen [24]. Dette betyr at tenner med lengst mineraliseringsfaser kan ha mer uttalte emaljedefekter enn tenner med kortere mineraliseringstid, og man tror at den autoimmune responsen mot emaljematriks i liten grad påvirker tannmorfologien etter at tennene er ferdig dannet [24].
Konklusjon
Pasienter med APS-1 har ofte orale manifestasjoner, og livslang oppfølging er viktig for å redusere plager og oppdage nye sykdomsmanifestasjoner tidlig. Et tverrfaglig samarbeid med fastleger og spesialisthelsetjenesten er essensielt for fullstendig diagnostisering, oppfølging og rett behandling.
Etikk
Studien er en del av et stort prosjekt på APS-1, som er godkjent av regional etisk komité (REK) Vest med nummer 2009_2555. Studier på oral helse ved APS-1 er også godkjent av REK Vest, med nummer 2020_212734. Pasientene, med bilder, som er beskrevet som kasus, har samtykket til studier på APS-I og spesifikt til gjengivelse i dette arbeidet.
Takk
Takk til pasienter som har sagt seg villig til å bidra til artikkelen med foto og sykdomshistorikk ved Tannhelsetjenestens kompetansesenter Vestland og Institutt for klinisk odontologi, Universitetet i Bergen.
Appendiks 1 oppsummerer anbefalinger om APS-1 for tannleger.
Appendiks 2 angir et skjema som er et hjelpemiddel for undersøkelse av pasienter med APS-1 eller som er under utredning for APS-1. Utarbeidet av Ivar Espelid, 2004. Samtykke til publisering er gitt. Oppdatert 2025, Universitetet i Oslo.
Referanser
Perheentupa J. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Clin Endocrinol Metab. 2006;91(8):2843–50.
Husebye ES, Anderson MS, Kampe O. Autoimmune Polyendocrine Syndromes. N Engl J Med. 2018;378(12):1132–41.
Wolff AS, Erichsen MM, Meager A, Magitta NF, Myhre AG, Bollerslev J, et al. Autoimmune polyendocrine syndrome type 1 in Norway: phenotypic variation, autoantibodies, and novel mutations in the autoimmune regulator gene. J Clin Endocrinol Metab. 2007;92(2):595–603.
Rosatelli MC, Meloni A, Devoto M, Cao A, Scott HS, Peterson P, et al. A common mutation in Sardinian autoimmune polyendocrinopathy- candidiasis-ectodermal dystrophy patients. Hum Genet. 1998;103(4):428–34.
Zlotogora J, Shapiro MS. Polyglandular autoimmune syndrome type I among Iranian Jews. J Med Genet. 1992;29(11):824–6.
Orlova EM, Bukina AM, Kuznetsova ES, Kareva MA, Zakharova EU, Peterkova VA, et al. Autoimmune polyglandular syndrome type 1 in Russian patients: clinical variants and autoimmune regulator mutations. Horm Res Paediatr. 2010;73(6):449–57.
Meloni A, Willcox N, Meager A, Atzeni M, Wolff AS, Husebye ES, et al. Autoimmune polyendocrine syndrome type 1: an extensive longitudinal study in Sardinian patients. J Clin Endocrinol Metab. 2012;97(4):1114–24.
Betterle C, Dalpra C, Greggio N, Volpato M, Zanchetta R. Autoimmunity in isolated Addison's disease and in polyglandular autoimmune diseases type 1, 2 and 4. Ann Endocrinol (Paris). 2001;62(2):193–201.
Betterle C, Greggio NA, Volpato M. Clinical review 93: Autoimmune polyglandular syndrome type 1. J Clin Endocrinol Metab. 1998;83(4):1049–55.
Bruserud O, Oftedal BE, Landegren N, Erichsen MM, Bratland E, Lima K, et al. A Longitudinal Follow-up of Autoimmune Polyendocrine Syndrome Type 1. J Clin Endocrinol Metab. 2016;101(8):2975–83.
An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. The Finnish-German APECED Consortium. Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy. Nat Genet. 1997;17(4):399–403.
Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, et al. Positional cloning of the APECED gene. Nat Genet. 1997;17(4):393–8.
Oftedal BE, Assing K, Baris S, Safgren SL, Johansen IS, Jakobsen MA, et al. Dominant-negative heterozygous mutations in AIRE confer diverse autoimmune phenotypes. iScience. 2023;26(6):106818.
Oftedal BE, Hellesen A, Erichsen MM, Bratland E, Vardi A, Perheentupa J, et al. Dominant Mutations in the Autoimmune Regulator AIRE Are Associated with Common Organ-Specific Autoimmune Diseases. Immunity. 2015;42(6):1185–96.
Oftedal BE, Wolff AS, Bratland E, Kampe O, Perheentupa J, Myhre AG, et al. Radioimmunoassay for autoantibodies against interferon omega; its use in the diagnosis of autoimmune polyendocrine syndrome type I. Clin Immunol. 2008;129(1):163–9.
Bruserud O, Oftedal BE, Wolff AB, Husebye ES. AIRE-mutations and autoimmune disease. Curr Opin Immunol. 2016;43:8–15.
Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298(5597):1395–401.
Husebye ES, Perheentupa J, Rautemaa R, Kampe O. Clinical manifestations and management of patients with autoimmune polyendocrine syndrome type I. J Intern Med. 2009;265(5):514–29.
Pavlic A, Waltimo-Siren J. Clinical and microstructural aberrations of enamel of deciduous and permanent teeth in patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Arch Oral Biol. 2009;54(7):658–65.
Ferre EM, Rose SR, Rosenzweig SD, Burbelo PD, Romito KR, Niemela JE, et al. Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI Insight. 2016;1(13).
McGovern E, Fleming P, Costigan C, Dominguez M, Coleman DC, Nunn J. Oral health in Autoimmune Polyendocrinopathy Candidiasis Ectodermal Dystrophy (APECED). Eur Arch Paediatr Dent. 2008;9(4):236–44.
Ferre EMN, Schmitt MM, Lionakis MS. Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy. Front Pediatr. 2021;9:723532.
Sveinsdóttir EG, Espelid, I. Dentale utviklingsforstyrrelser: kliniske utfordringer i diagnostikk og behandling. Aktuel Nordisk Odontologi. 2016;41(1):126–45.
Gruper Y, Wolff ASB, Glanz L, Spoutil F, Marthinussen MC, Osickova A, et al. Autoimmune amelogenesis imperfecta in patients with APS-1 and coeliac disease. Nature. 2023;624(7992):653–62.
Humbert L, Cornu M, Proust-Lemoine E, Bayry J, Wemeau JL, Vantyghem MC, et al. Chronic Mucocutaneous Candidiasis in Autoimmune Polyendocrine Syndrome Type 1. Front Immunol. 2018;9:2570.
McManus BA, McGovern E, Moran GP, Healy CM, Nunn J, Fleming P, et al. Microbiological screening of Irish patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy reveals persistence of Candida albicans strains, gradual reduction in susceptibility to azoles, and incidences of clinical signs of oral candidiasis without culture evidence. J Clin Microbiol. 2011;49(5):1879–89.
Bruserud O, Costea DE, Laakso S, Garty BZ, Mathisen E, Makitie A, et al. Oral Tongue Malignancies in Autoimmune Polyendocrine Syndrome Type 1. Front Endocrinol (Lausanne). 2018;9:463.
Ahonen P, Myllarniemi S, Sipila I, Perheentupa J. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med. 1990;322(26):1829–36.
Tang XH, Urvalek AM, Osei-Sarfo K, Zhang T, Scognamiglio T, Gudas LJ. Gene expression profiling signatures for the diagnosis and prevention of oral cavity carcinogenesis-genome-wide analysis using RNA-seq technology. Oncotarget. 2015;6(27):24424–35.
Zhang HX, Liu OS, Deng C, He Y, Feng YQ, Ma JA, et al. Genome-wide gene expression profiling of tongue squamous cell carcinoma by RNA-seq. Clin Oral Investig. 2018;22(1):209–16.
Oftedal BE, Marthinussen MC, Erichsen MM, Tveitaras MK, Kjellesvik-Kristiansen A, Hammenfors D, et al. Impaired salivary gland activity in patients with autoimmune polyendocrine syndrome type I. Autoimmunity. 2017;50(4):211–22.
Løkken P, Birkeland JM. Medikamenter og munntørrhet. Tidsskr Nor Lægeforen. 2005;125(5):581–2.
Oftedal BE HES. Autoimmune polyglandulære syndromer – er monogene former vanligere enn vi tror? . Indremedisineren. 2016;3:14–7.
Helsedirektoratet. SMT-listen [nettdokument] Helsedirektoratet2020 [oppdatert 01.02.2025.] Tilgjengelig fra: https://www.helsedirektoratet.no/rundskriv/folketrygdloven-kap-5/folketrygdloven--5-6--5-6-a-og--5-25--undersokelse-og-behandling-hos-tannlege-og-tannpleier-for-sykdom-og-skade/smt-listen.
TAKO-senteret LDS. Autoimmunt polyendokrint syndrom 1 (APS type 1) 2023 [oppdatert 03.07.2025.] Tilgjengelig fra: https://www.lovisenbergsykehus.no/tako-senteret/autoimmunt-polyendokrint-syndrom-type-1-aps-type-1.
Eidt G, Waltermann EDM, Hilgert JB, Arthur RA. Candida and dental caries in children, adolescents and adults: A systematic review and meta-analysis. Arch Oral Biol. 2020;119:104876.
Stoltze K. Bivirkninger af natriumlaurylsulfat i mundplejemidler? Tandlaegebladet. 2006;5(104).
Astor MC, Lovas K, Debowska A, Eriksen EF, Evang JA, Fossum C, et al. Epidemiology and Health-Related Quality of Life in Hypoparathyroidism in Norway. J Clin Endocrinol Metab. 2016;101(8):3045–53.
Ponranjini VC, Jayachandran S, Kayal L, Bakyalakshmi K. Autoimmune polyglandular syndrome type 1. J Clin Imaging Sci. 2012;2:62.
Wang NJ. Oversikt over vanlige mineraliseringsforstyrrelser i emaljen og erfaringer i klinikken. Nor Tannlegeforen Tid. 2016;126:764–70.
Afzal SH, Skaare AB, Wigen TI, Brusevold IJ. Molar-Incisor Hypomineralisation: Severity, caries and hypersensitivity. J Dent. 2024;142:104881.
English summary
Oral manifestations of autoimmune polyendrocrine syndrome type 1
Autoimmune polyendocrine syndrome type 1 (APS-1) is a rare autoimmune disease characterized by primary adrenal insufficiency (Addison’s disease), chronic mucocutaneous candidiasis and hypoparathyroidism. Disease manifestations vary from individual to individual, and both endocrine and non-endocrine tissues are affected with debute in childhood, but often with delayed diagnosis. Oral manifestations as enamel defects and chronic oral candida infections are among the disease hallmarks. The enamel may be affected to various degrees within the same patient and between disease cases, from minor hypomineralization to serious enamel hypoplasia affecting most of the teeth. This is expected to be underdiagnosed and should be addressed on a larger scale when examining patients with this disease pattern. Chronic oral candidiasis from Candida albicans in oral mucosa and mucosa of the esophagus in APS-1 patients may develop over time to dysplastic changes in the mucosa.Ulceration and atrophic mucosa is furthermore commonly seen.
The oral disease hallmarks of APS-1 underline the important awareness by dental health care professionals of this syndrome. This will provide better diagnostics at an earlier stage of the disease and improved treatment by interdisciplinary collaborations between general practitioners, endocrinologists and dental health care providers.
Keywords: polyendocrinopathies, autoimmune, developmental defects of enamel, dental enamel hypoplasia
Nøkkelord: polyendokrinopatier, autoimmune sykdommer; utviklingsdefekter i emalje; hypoplasi av tannemalje
Akseptert for publisering 15.12.2025. Artikkelen er fagfellevurdert.
Artikkelen siteres som:
Borge H, Gjerde C, Bletsa A, Guddal E, Runningen A, Sivakumar A, Kvalheim SF, Nordgarden H, Skarstein K, Husebye ES, Wolff ASB, Marthinussen MC. Orale manifestasjoner ved autoimmunt polyendokrint syndrom type 1. Nor Tannlegeforen Tid. 2026;. doi:10.56373/6964ed27d4351